Curator’s guide¶
Please note that links displayed within the curation interface will vary depending on database contents and the permissions of the curator.
Adding new sender details¶
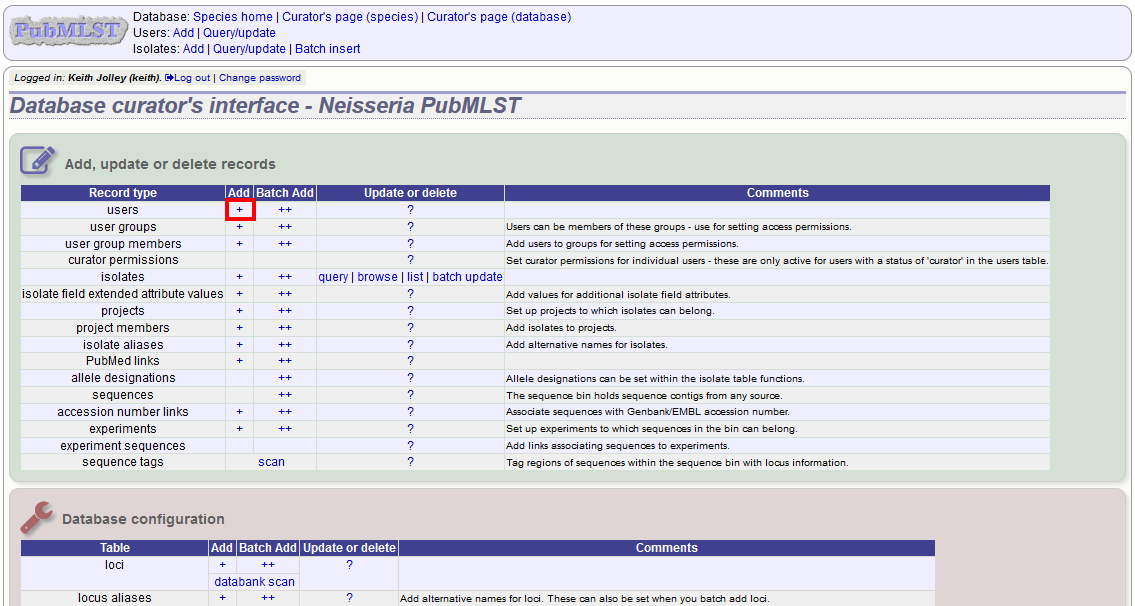
All records within the databases are associated with a sender. Whenever somebody new submits data, they should be added to the users table so that their name appears in the dropdown lists on the data upload forms.
To add a user, click the add users (+) link on the curator’s contents page.
Enter the user’s details in to the form.
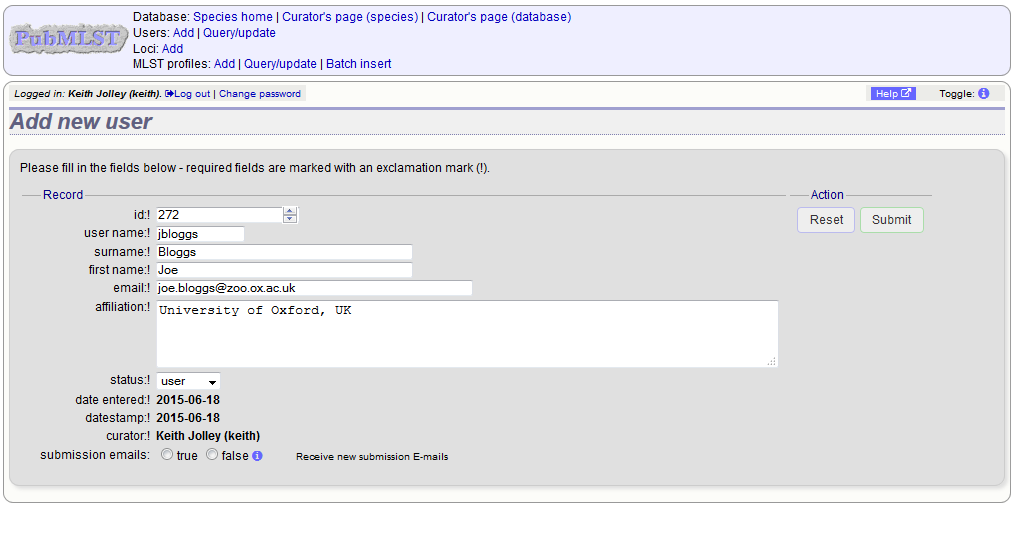
Normally the status should be set as ‘user’. Only admins and curators with special permissions can create users with a status of curator or admin.
If the submission system is in operation there will be an option at the bottom called ‘submission_emails’. This is to enable users with a status of ‘curator’ or ‘admin’ to receive E-mails on receipt of new submissions. It is not relevant for users with a status of ‘user’ or ‘submitter’.
Adding new allele sequence definitions¶
Single allele¶
To add a single new allele, click the sequences (all loci) add (+) link on the curator’s main page - if only a few loci are defined with permission for the current user to curate then they will be listed individually and the specific locus allele addition links can also be used.
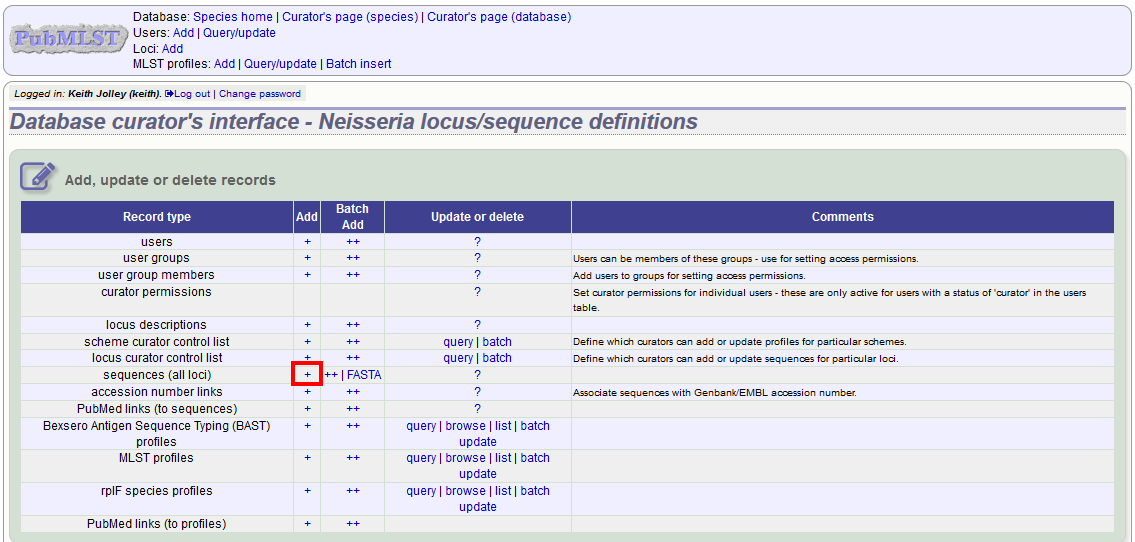
Select the locus from the dropdown list box. The next available allele id will be entered automatically (if the allele id format is set to integer). Paste the sequence in to form, set the status and select the sender name from the dropdown box. If the sender does not appear in the box, you will need to add them to the registered users.
The status reflects the level of curation that the curator has done personally - the curator should not rely on assurances from the submitter. The status can either be:
- Sanger trace checked
- Sequence trace files have been assembled and inspected by the curator.
- WGS: manual extract (BIGSdb)
- The sequence has been extracted manually from a BIGSdb database by the curator . There may be some manual intervention to identify the start and stop sites of the sequence.
- WGS: automated extract (BIGSdb)
- The sequences have been generated by a BIGSdb tag scanning run and have had no manual inspection or intervention.
- WGS: visually checked
- Short read data has been inspected visually using an alignment program by the curator.
- WGS: automatically checked
- The sequences have been checked by an automated algorithm that assesses the quality of the data to ensure it meets specified criteria.
- unchecked
- If none of the above match, then the sequence should be entered as unchecked.
You can also choose whether to designate the sequence as a type allele or not. Type alleles can be used to constrain the sequence search space when defining new alleles using the web-based scanner or offline auto allele definer.
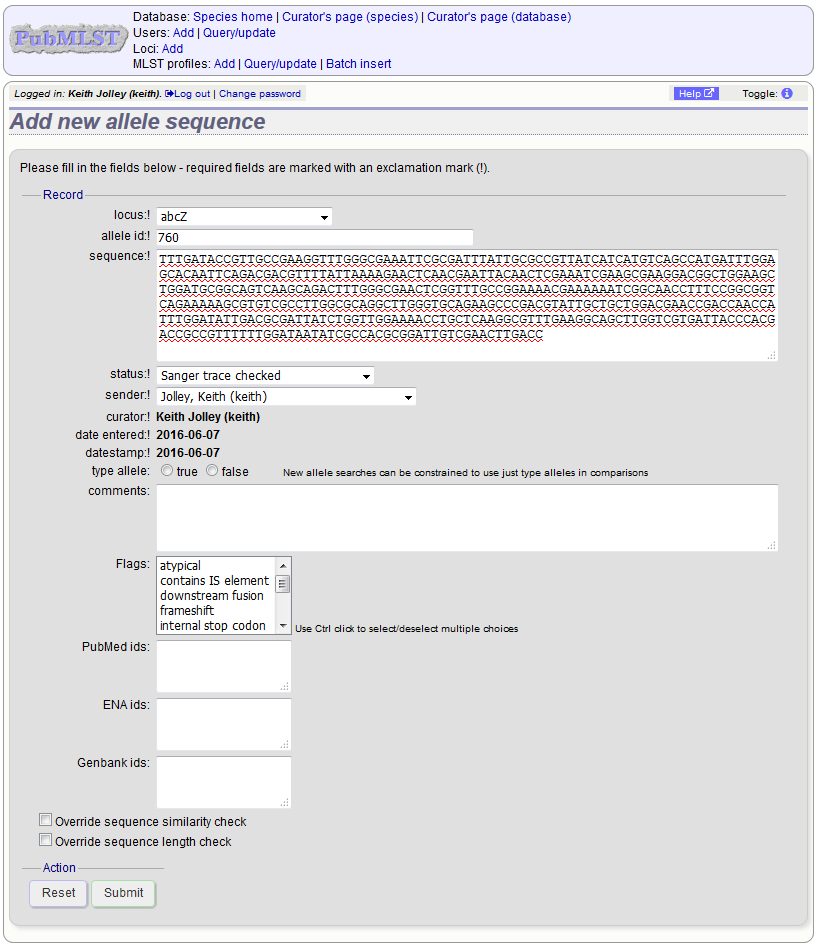
Press submit. By default, the system will test whether your sequence is similar enough to existing alleles defined for that locus. The sequence will be rejected if it isn’t considered similar enough. This test can be overridden by checking the ‘Override sequence similarity check’ checkbox at the bottom. It will also check that the sequence length is within the allowed range for that locus. These checks can also be overridden by checking the ‘Override sequence length check’ checkbox, allowing the addition of unusual length alleles.
See also
Sequences can also be associated with PubMed, ENA or Genbank id numbers by entering these as lists (one value per line) in the appropriate form box.
Batch adding multiple alleles¶
There are two methods of batch adding alleles. You can either upload a spreadsheet with all fields in tabular format, or you can upload a FASTA file provided all sequences are for the same locus and have the same status.
Upload using a spreadsheet¶
Click the batch add (++) sequences (all loci) link on the curator’s main page.
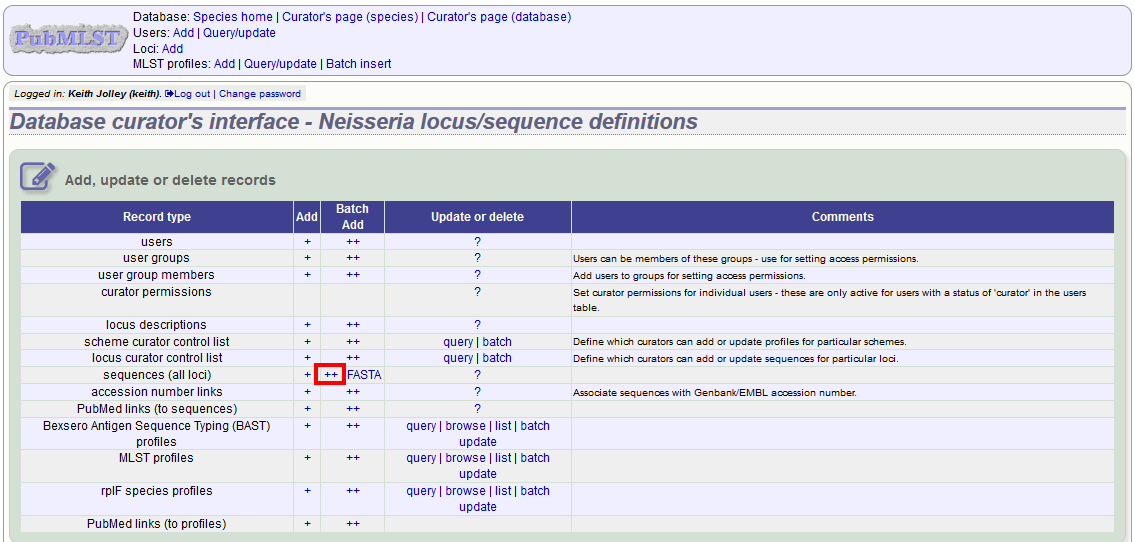
Download a template Excel file from the following page.
Fill in the spreadsheet. If the locus uses integer allele identifiers, the allele_id can be left blank and the next available number will be used automatically.
The status can be either: ‘Sanger trace checked’, ‘WGS: manual extract (BIGSdb)’, ‘WGS: automated extract (BIGSdb)’, ‘WGS: visually checked’, ‘WGS: automatically checked’ or ‘unchecked’. See full explanations for these in the single allele upload section.
The ‘type_allele’ field is boolean (true/false) and specifies if the sequence should be considered as a type allele. These can be used to constrain the sequence search space when defining new alleles using the web-based scanner or offline auto allele definer.
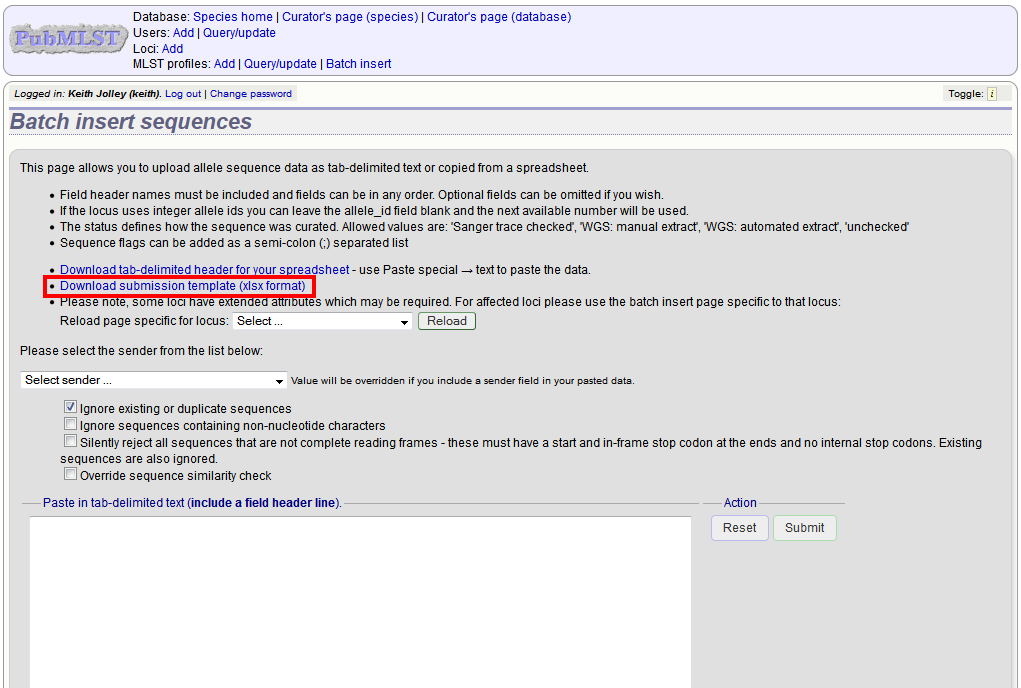
Paste the entire sheet in to the web form and select the sender from the dropdown box.
Additionally, there are a number of options available. Some of these will ignore sequences if they don’t match certain criteria - this is useful when sequence data has been extracted from genomes automatically. Available options are:
- Ignore existing or duplicate sequences.
- Ignore sequences containing non-nucleotide characters.
- Silently reject all sequences that are not complete reading frames - these must have a start and in-frame stop codon at the ends and no internal stop codons. Existing sequences are also ignored.
- Override sequence similarity check.
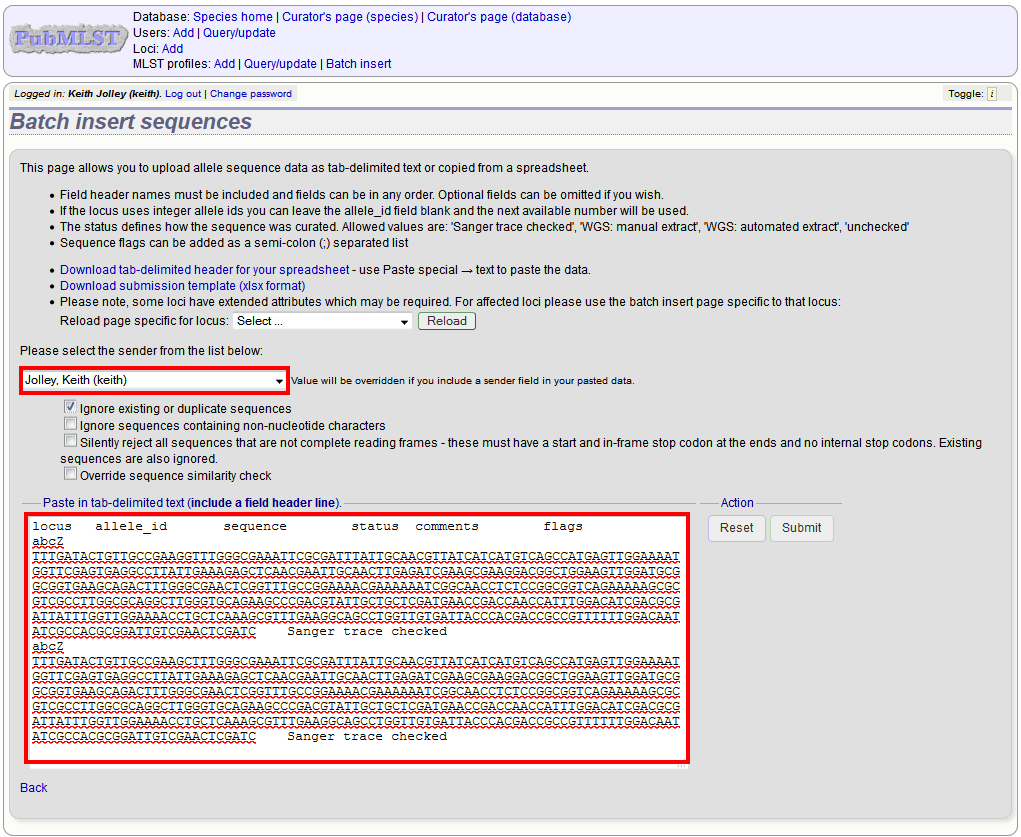
Press submit. You will be presented with a page indicating what data will be uploaded. This gives you a chance to back out of the upload. Click ‘Import data’.
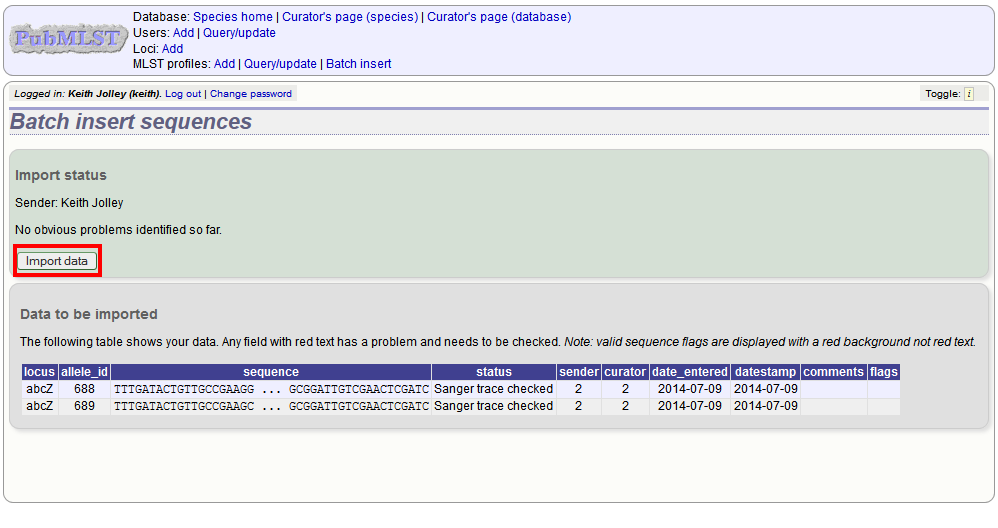
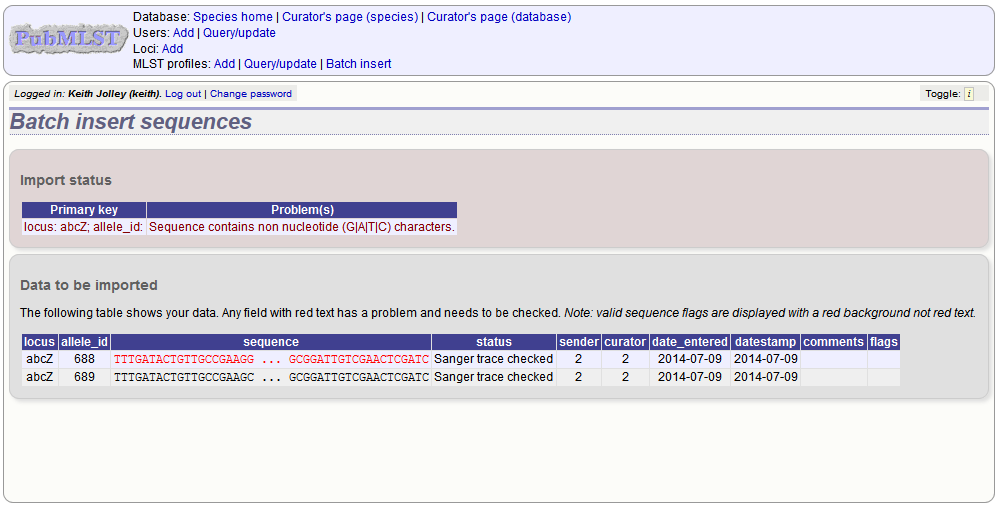
If there are any problems with the submission, these should be indicated at this stage, e.g.:
Upload using a FASTA file¶
Uploading new alleles from a FASTA file is usually more straightforward than generating an Excel sheet.
Click ‘FASTA’ upload on the curator’s contents page.
Select the locus, status and sender from the dropdown boxes and paste in the new sequences in FASTA format.
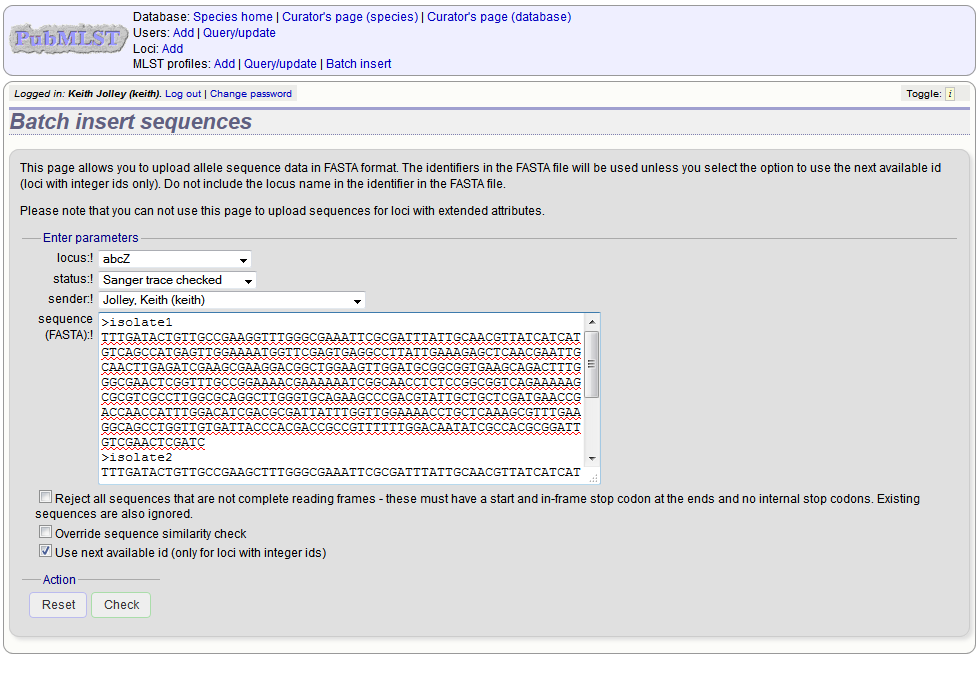
For loci with integer ids, the next available id number will be used by default (and the identifier in the FASTA file will be ignored). Alternatively, you can indicate the allele identifier within the FASTA file (do not include the locus name in this identifier).
As with the spreadsheet upload, you can select options to ignore selected sequences if they don’t match specific criteria.
Click ‘Check’.
The sequences will be checked. You will be presented with a page indicating what data will be uploaded. This gives you a chance to back out of the upload. Click ‘Upload valid sequences’.
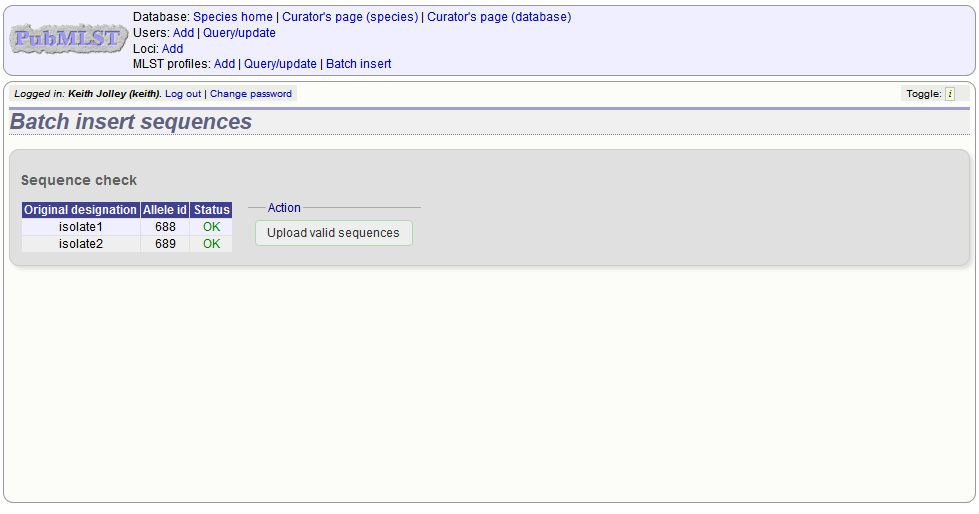
Any invalid sequences will be indicated in this confirmation page and these will not be uploaded (you can still upload the others), e.g.
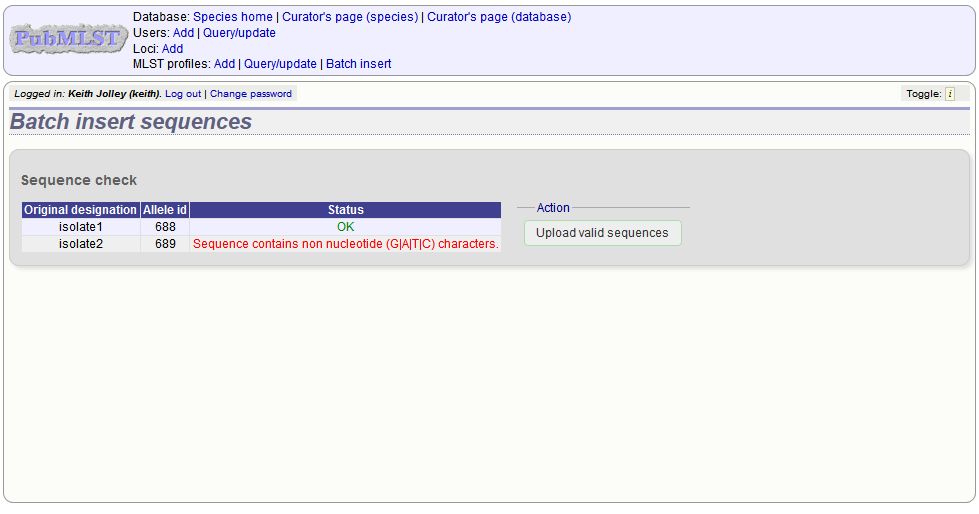
Updating and deleting allele sequence definitions¶
Note
You cannot update the sequence of an allele definition. This is for reasons of data integrity since an allele may form part of a scheme profile and be referred to in multiple databases. If you really need to change a sequence, you will have to remove the allele definition and then re-add it. If the allele is a member of a scheme profile, you will also have to remove that profile first, then re-create it after deleting and re-adding the allele.
In order to update or delete an allele, first you must select it. Click the query (?) sequences (all loci) link - if only a few loci are defined with permission for the current user to curate then they will be listed individually and the specific locus query links can also be used.
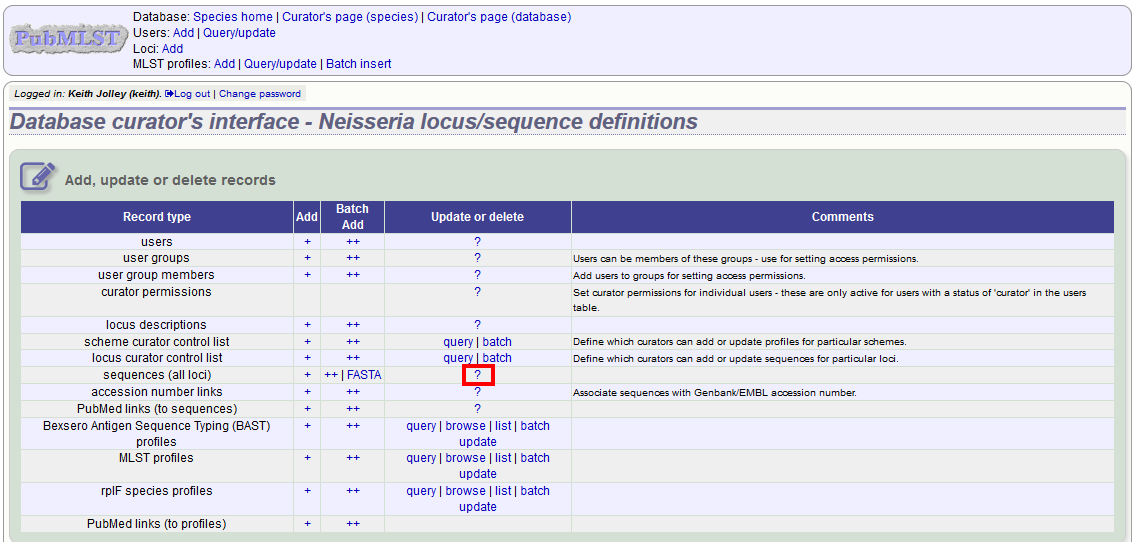
Either search for specific attributes in the search form, or leave it blank and click ‘Submit’ to return all alleles. For a specific allele, select the locus in the filter and enter the allele number in the allele_id field.
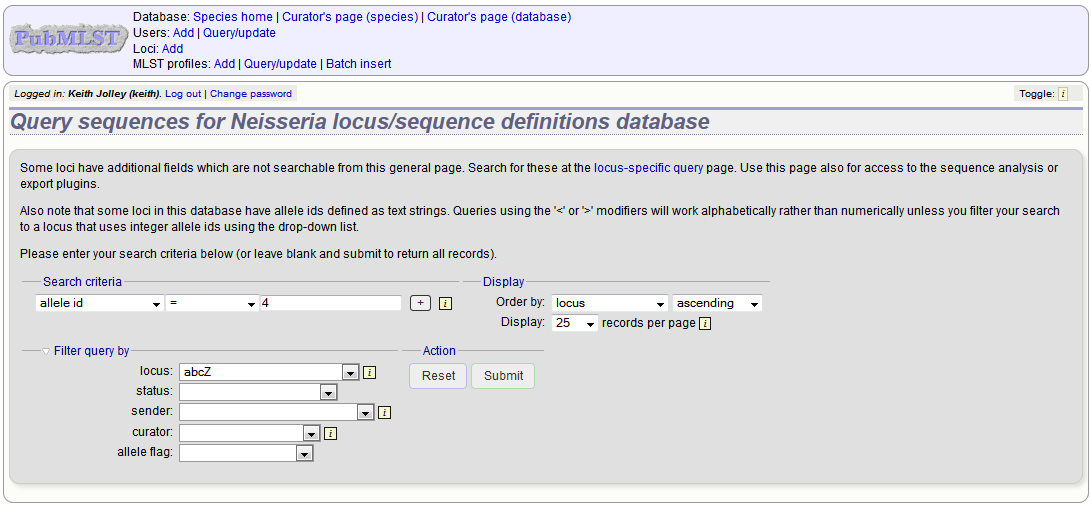
Click the appropriate link to either update the allele attributes or to delete it. If you have appropriate permissions, there may also be a link to ‘Delete ALL’. This allows you to quickly delete all alleles returned from a search.
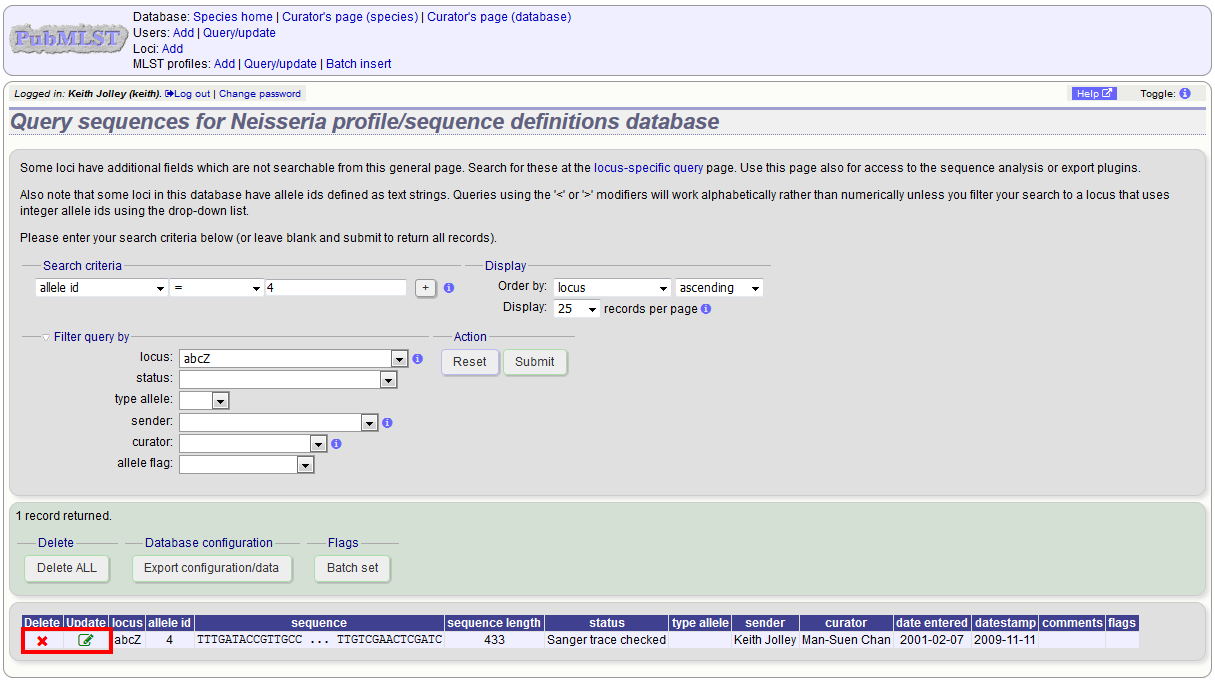
If you choose to delete, you will be presented with a final confirmation screen. To go ahead, click ‘Delete!’. Deletion will not be possible if the allele is part of a scheme profile - if it is you will need to delete any profiles that it is a member of first. You can also choose to delete and retire the allele identifier. If you do this, the allele identifier will not be re-used.
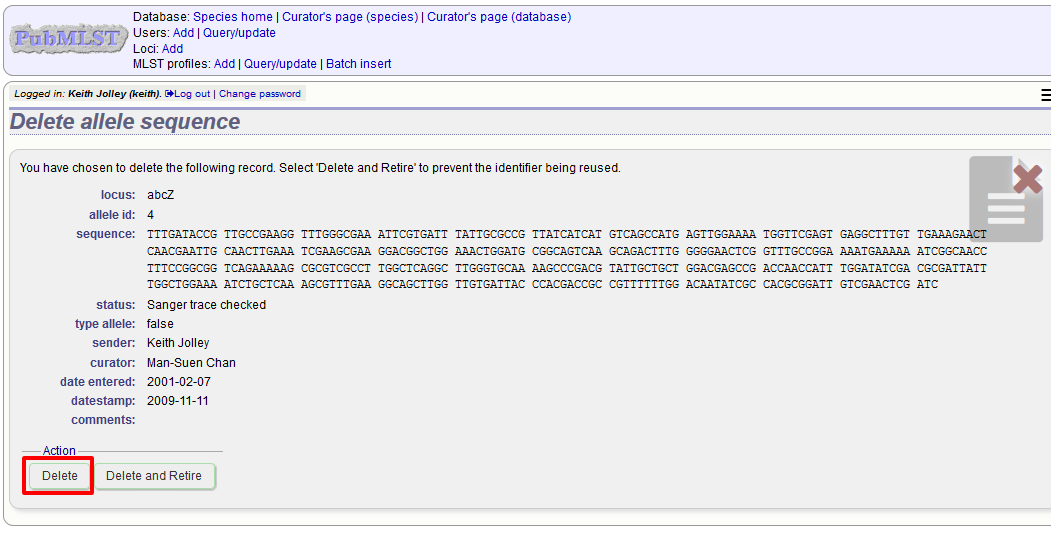
If instead you clicked ‘Update’, you will be able to modify attributes of the sequence, or link PubMed, ENA or Genbank records to it. You will not be able to modify the sequence itself.
Note
Adding flags and comments to an allele record requires that this feature is enabled in the database configuration.
Retiring allele identifiers¶
Sometimes there is a requirement to prevent the automated assignment of a particular allele identifier - an allele with that identifier may have been commonly used and has since been removed. Reassignment of the identifier to a new sequence may lead to confusion, so in this instance, it would be better to prevent this.
You can retire an allele identifier by clicking the ‘Add’ retired allele ids link on the sequence database curators’ page.
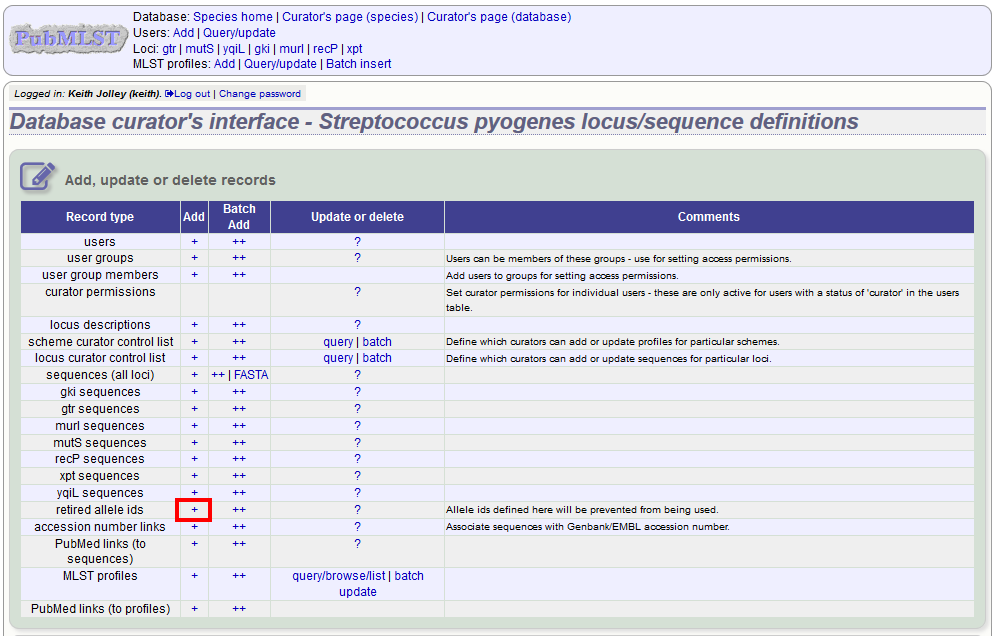
Select the locus from the dropdown list box and enter the allele id. Click ‘Submit’.
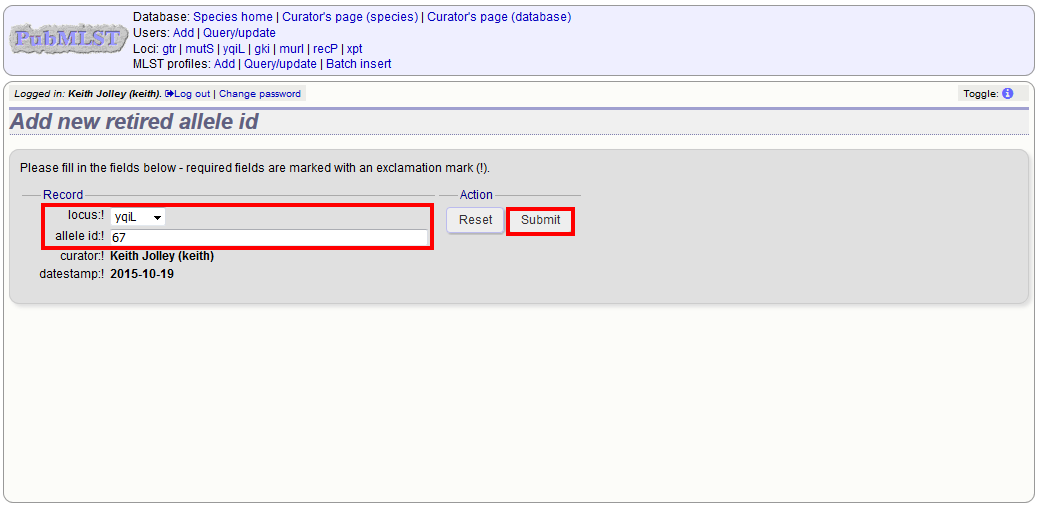
You cannot retire an allele that already exists, so you must delete it before retiring it. Once an identifier is retired, you will not be able to create a new allele with that name.
You can also retire an allele identifier when you delete an allele.
Updating locus descriptions¶
Loci in the sequence definitions database can have a description associated with them. This may contain information about the gene product, the biochemical reaction it catalyzes, or publications providing more detailed information etc. This description is accessible from various pages within the interface such as an allele information page or from the allele download page.
Note
In recent versions of BIGSdb, a blank description record is created when a new locus is defined. The following instructions assume that this is the case. It is possible for this record to be deleted or it may never have existed if the locus was created using an old version of BIGSdb. If the record does not exist, it can be added by clicking the Add (+) button next to ‘locus descriptions’. Fill in the fields in the same way as described below.
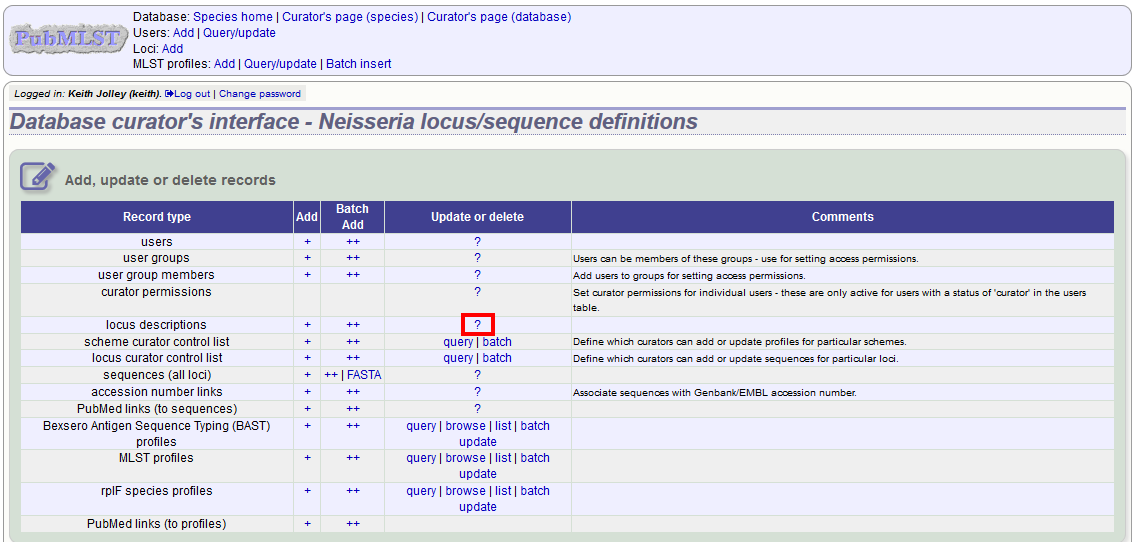
To edit a locus description, first you need to find it. Click the update/delete (?) button next to ‘locus descriptions’ on the sequence database curator’s page (depending on the permissions set for your user account not all the links shown here may be displayed).
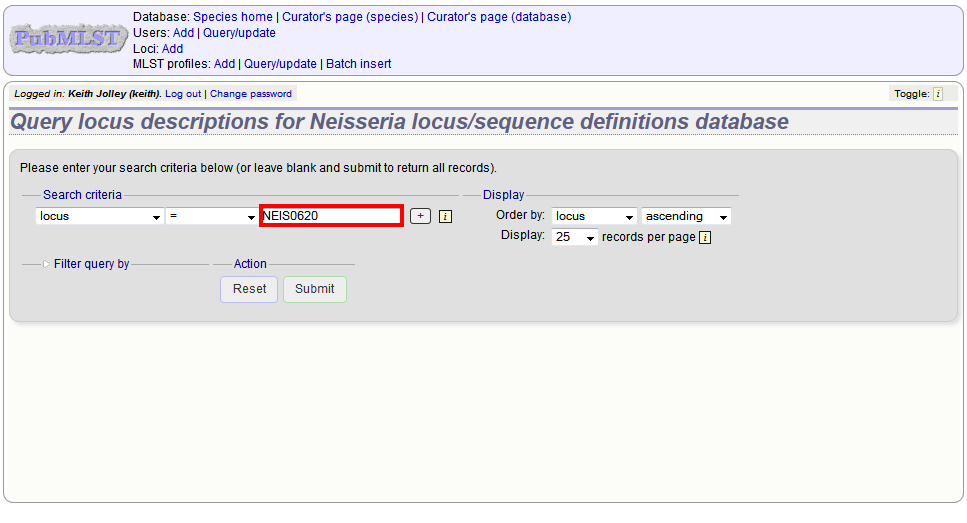
Either enter the name of the locus in the query box:
or expand the filter list and select it from the dropdown box:
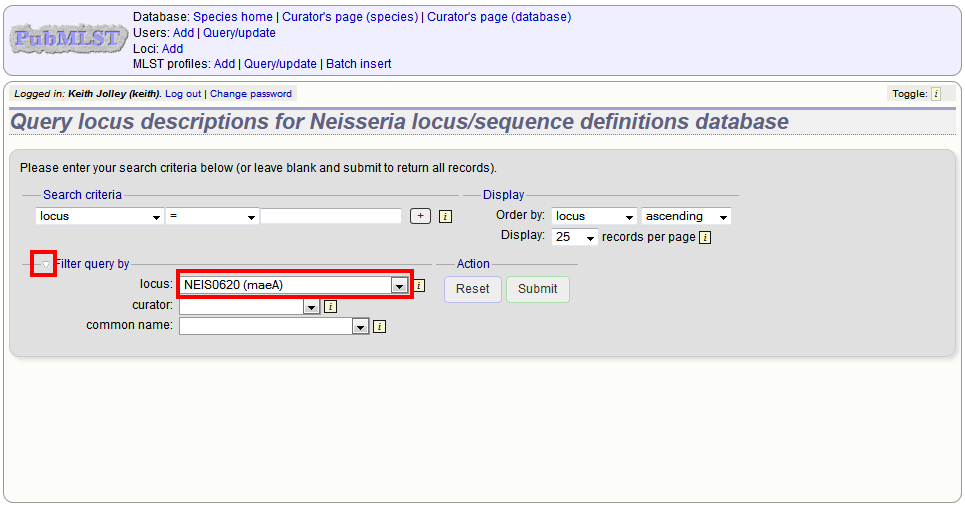
Click ‘Submit’.
If the locus description exists, click the ‘Update’ link (if it doesn’t, see the note above).
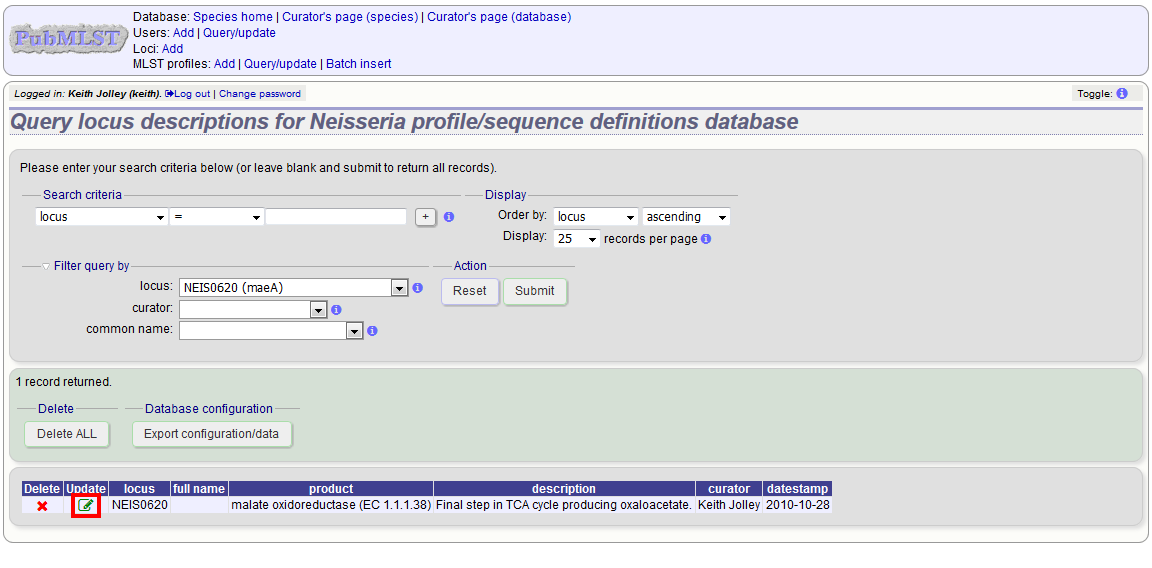
Fill in the form as needed:
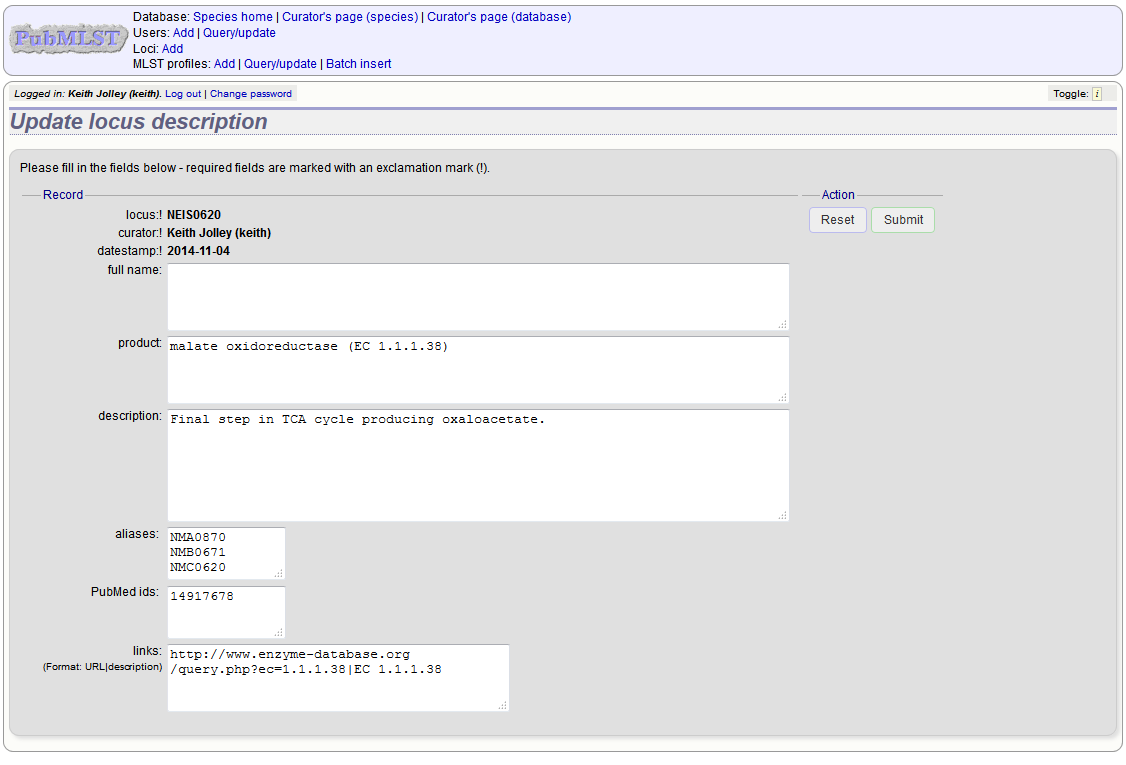
full_name
The full name of the locus - often this can be left blank as it may be the same as the locus name. An example of where it is appropriately used is where the locus name is an abbreviation, e.g. PorA_VR1 - here we could enter ‘PorA variable region 1’. This should not be used for the ‘common name’ of the locus (which is defined within the locus record itself) or the gene product.
product
The name of the protein product of a coding sequence locus.
description
This can be as full a description as possible. It can include the specific part of the biochemical pathway the gene product catalyses or may provide background information, as appropriate.
aliases
These are alternative names for the locus as perhaps found in different genome annotations. Don’t duplicate the locus name or common name defined in the locus record. Enter each alias on a separate line.
Pubmed_ids
Enter the PubMed id of any paper that specifically describes the locus. Enter each id on a separate line. The software will retrieve the full citation from PubMed (this happens periodically so it may not be available for display immediately).
Links
Enter links to additional web-based resources. Enter the URL first followed by a pipe symbol (|) and then the description.
Click ‘Submit’ when finished.
Adding new scheme profile definitions¶
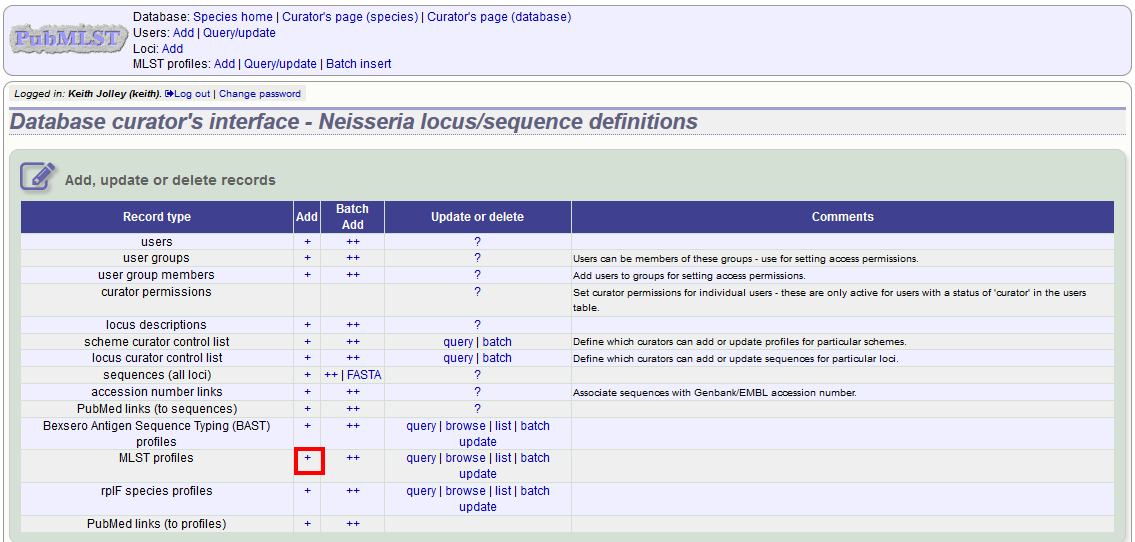
Provided a scheme has been set up with at least one locus and a scheme field set as a primary key, there will be links on the curator’s main page to add profiles for that scheme.
To add a single profile you can click the add (+) profiles link next to the scheme name (e.g. MLST):
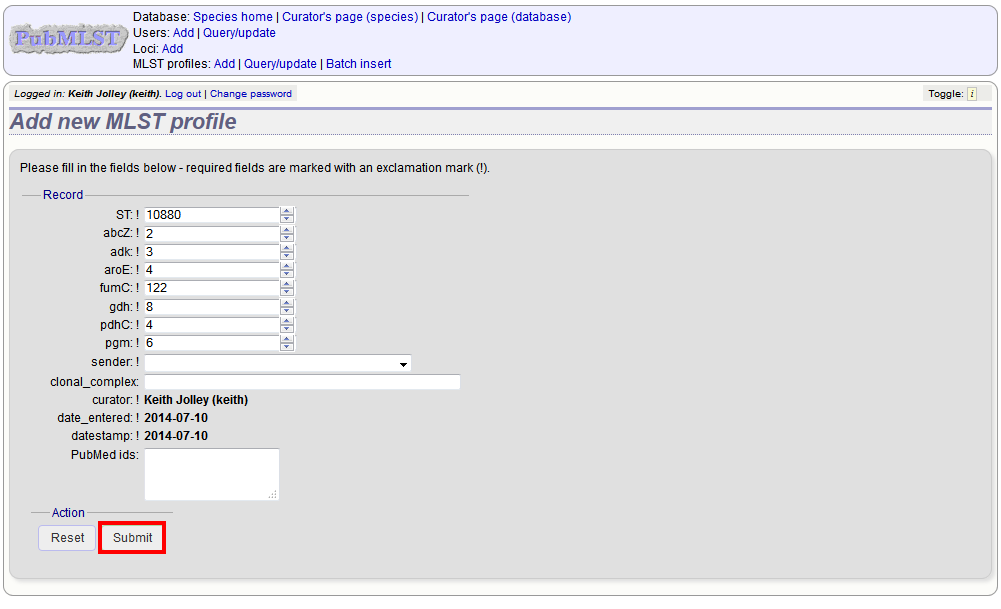
A form will be displayed with the next available primary key number already entered (provided integers are used for the primary key format). Enter the new profile, associated scheme fields, and the sender, then click ‘Submit’. The new profile will be added provided the primary key or the profile has not previously been entered.
More usually, profiles are added in a batch mode. It is often easier to do this even for a single profile since it allows copying and pasting data from a spreadsheet.
Click the batch add (++) profiles link next to the scheme name:
Click the ‘Download submission template (xlsx format)’ link to download an Excel submission template.
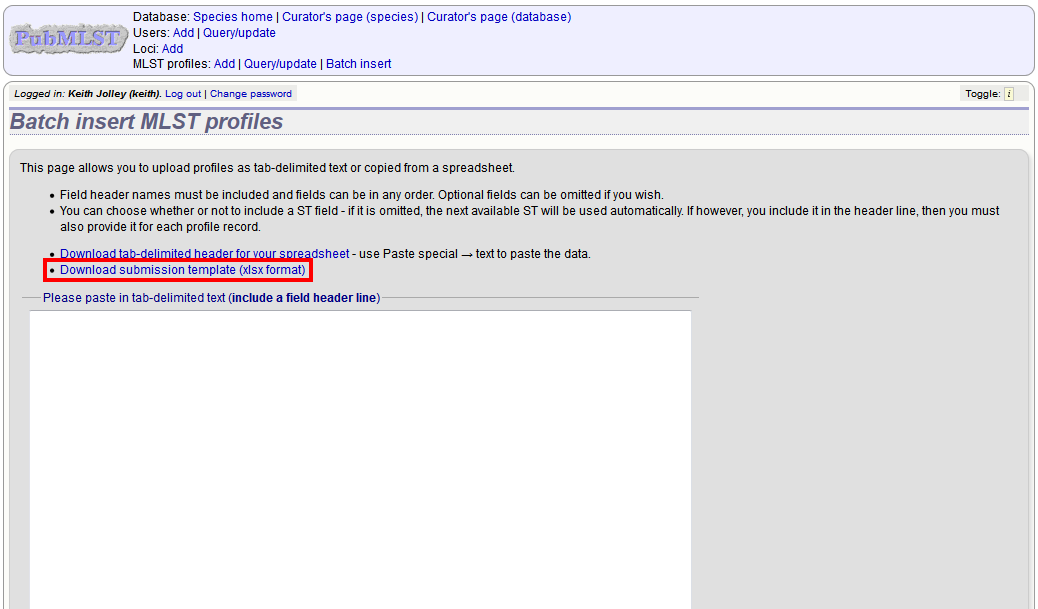
Fill in the spreadsheet using the copied template, then copy and paste the whole spreadsheet in to the large form on the upload page. If the primary key has an integer format, you can exclude this column and the next available number will be used automatically. If the column is included, however, a value must be set. Select the sender from the dropdown list box and then click ‘Submit’.
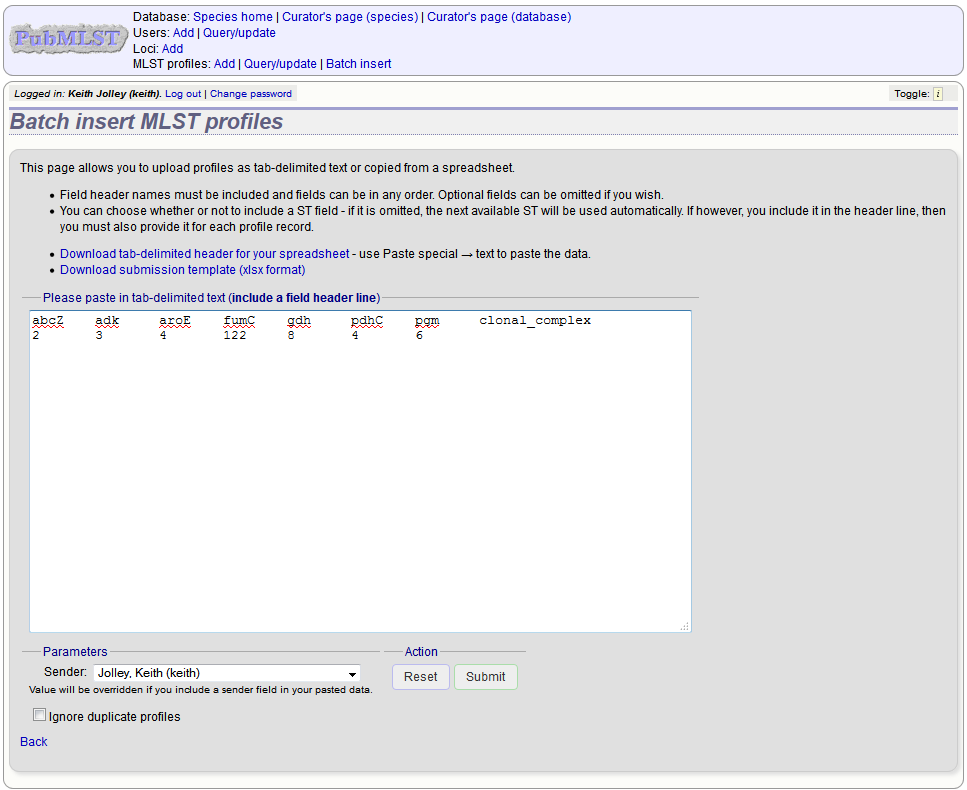
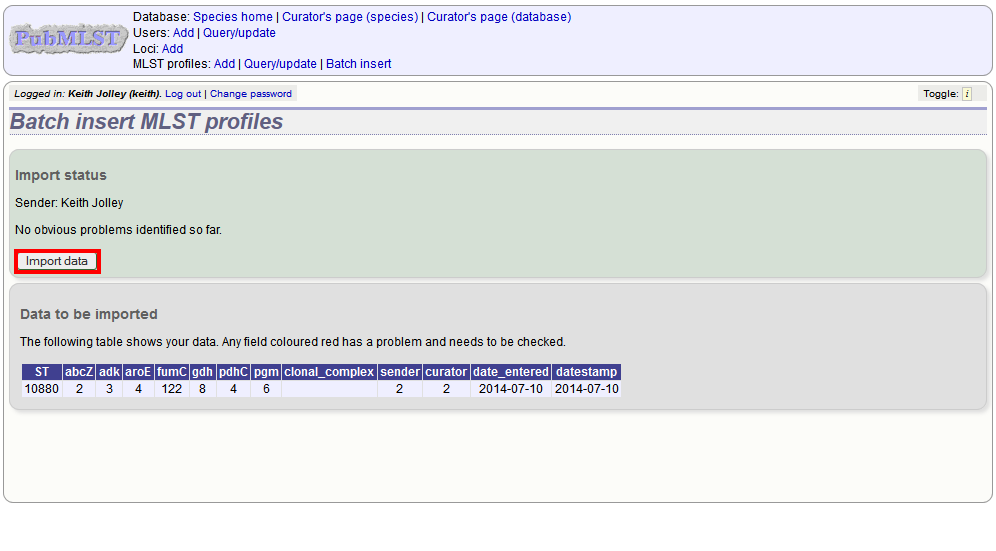
You will be given a final confirmation page stating what will be uploaded. If you wish to proceed with the submission, click ‘Import data’.
Updating and deleting scheme profile definitions¶
In order to update or delete a scheme profile, first you must select it. Click the query (?) profiles link next to the scheme name (e.g. MLST):
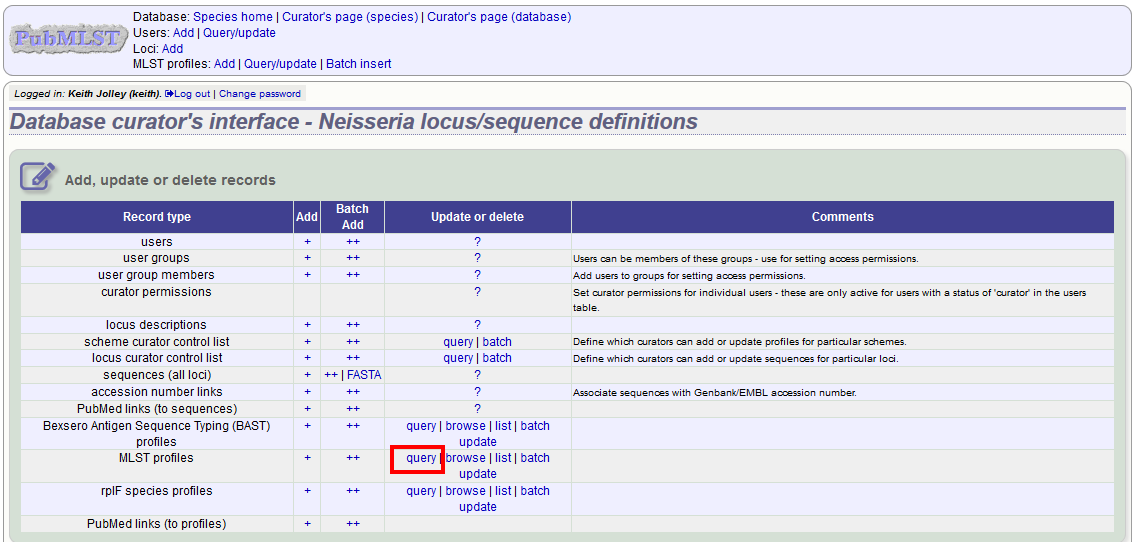
Search for your profile by entering search criteria (alternatively you can use the browse or list query functions).
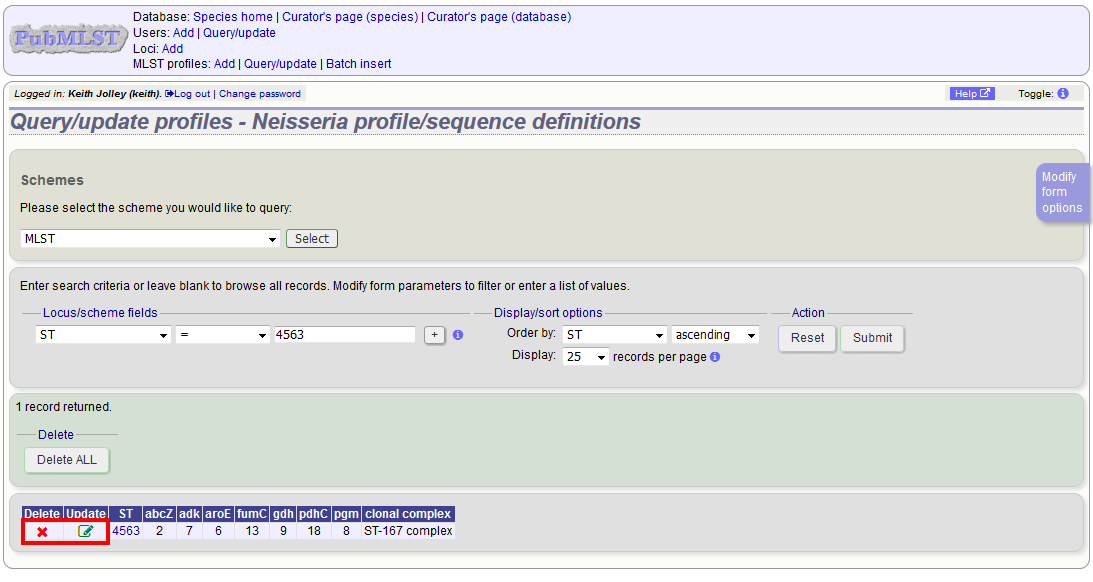
To delete the profile, click the ‘Delete’ link next to the profile. Alternatively, if your account has permission, you may be able to ‘Delete ALL’ records retrieved from the search.
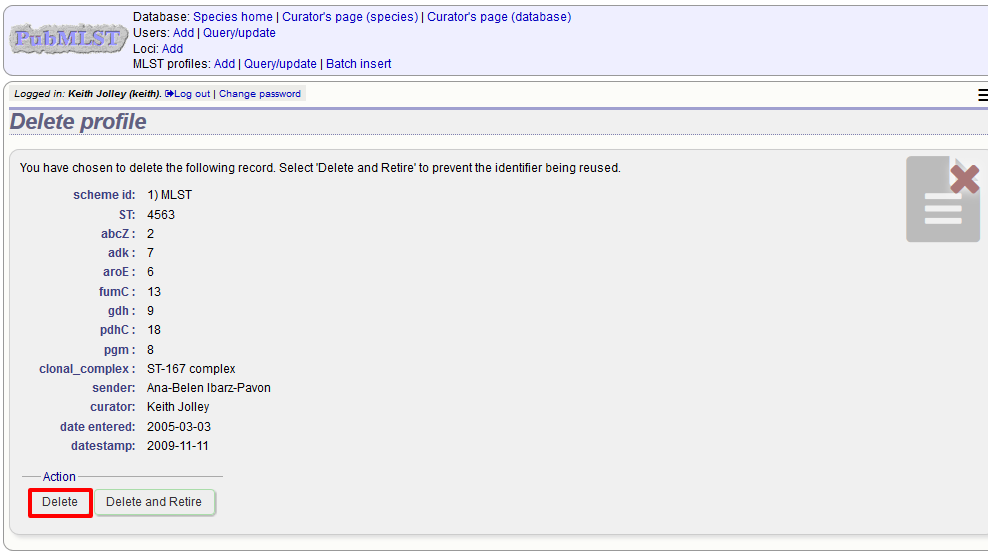
For deletion of a single record, the full record will be displayed. Confirm deletion by clicking ‘Delete’. You can also choose to delete and retire the profile identifier. If you do this, the profile identifier will not be re-used.
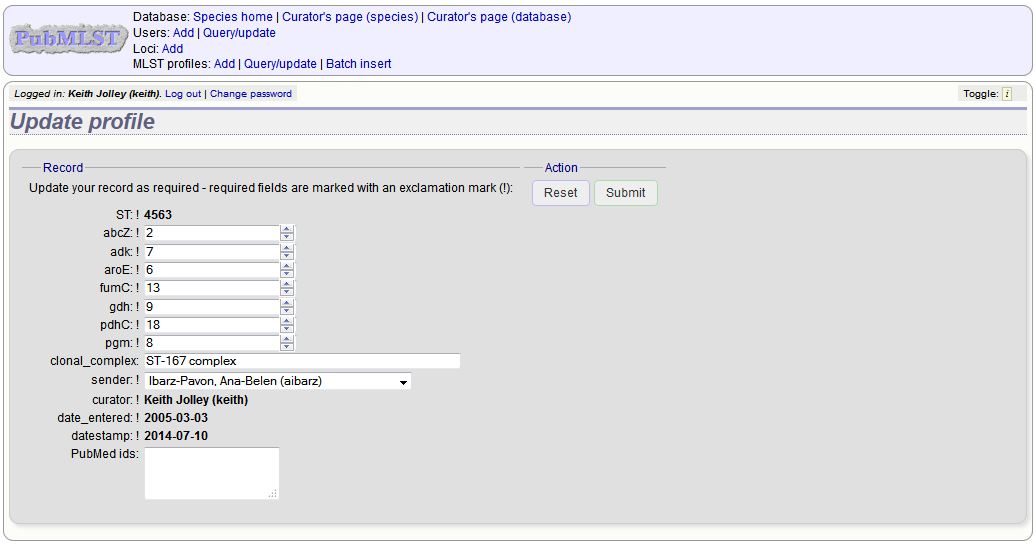
To modify the profile, click the ‘Update’ link next to the profile following the query. A form will be displayed - make any changes and then click ‘Update’.
Retiring scheme profile definitions¶
Sometimes there is a requirement to prevent the automated assignment of a particular profile identifier (e.g. ST) - a profile with that identifier may have been commonly used and has since been removed. Reassignment of the identifier to a new profile may lead to confusion, so in this instance, it would be better to prevent this.
You can retire a profile identifier by clicking the ‘Add’ retired profiles link on the sequence database curators’ page.
Select the scheme from the dropdown list box and enter the profile id. Click ‘Submit’.
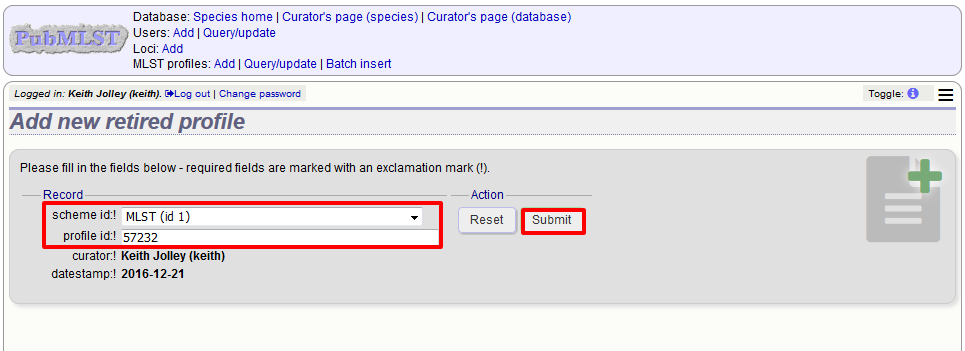
You cannot retire a profile identifier that already exists, so you must delete it before retiring it. Once an identifier is retired, you will not be able to create a new profile with that name.
You can also retire a profile definition when you delete a profile.
Adding isolate records¶
To add a single record, click the add (+) isolates link on the curator’s index page.
The next available id will be filled in automatically but you are free to change this. Fill in the individual fields. Required fields are listed first and are marked with an exclamation mark (!). Some fields may have drop-down list boxes of allowed values. You can also enter allele designations for any loci that have been defined.
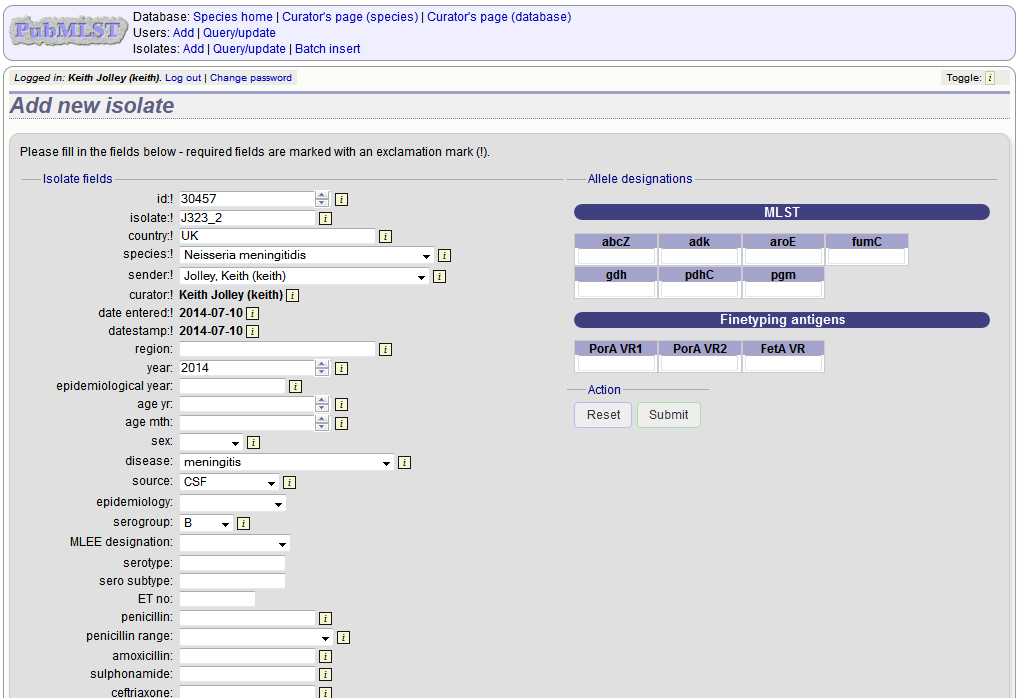
Press submit when finished.
More usually, isolate records are added in batch mode, even when only a single record is added, since the submission can be prepared in a spreadsheet and copied and pasted.
Select batch add (++) isolates link on the curator’s index page.
Download a submission template in Excel format from the link.
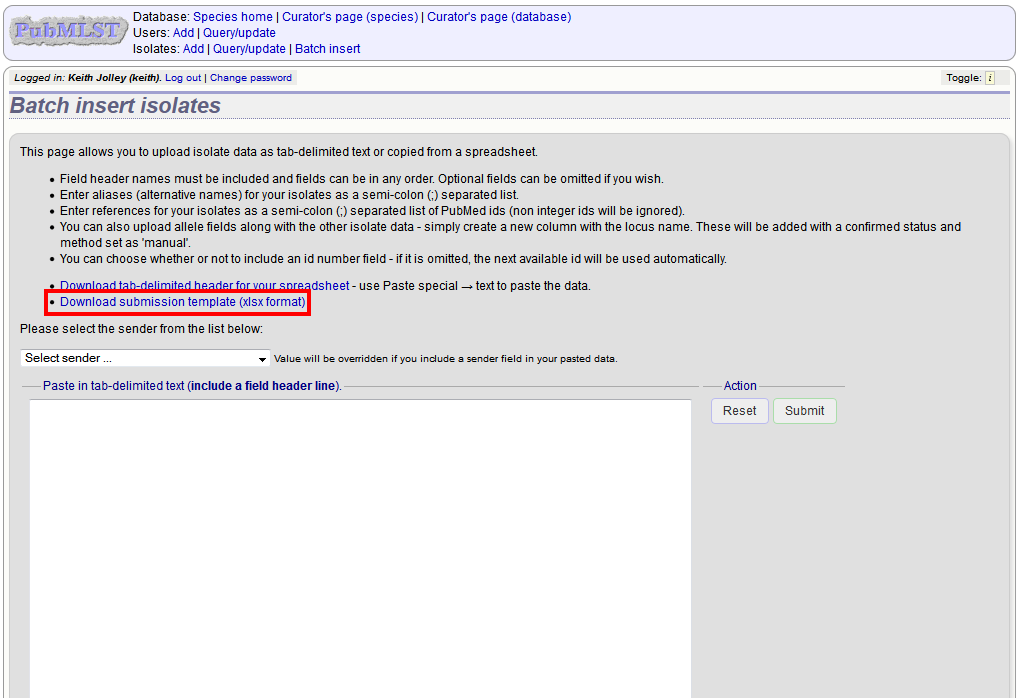
Prepare your data in the spreadsheet - the column headings must match the database fields. In databases with large numbers of loci, there won’t be columns for each of these. You can, however, manually add locus columns.
Pick a sender from the drop-down list box and paste the data from your spreadsheet in to the web form. The next available isolate id number will be used automatically (this can be overridden if you manually add an id column).
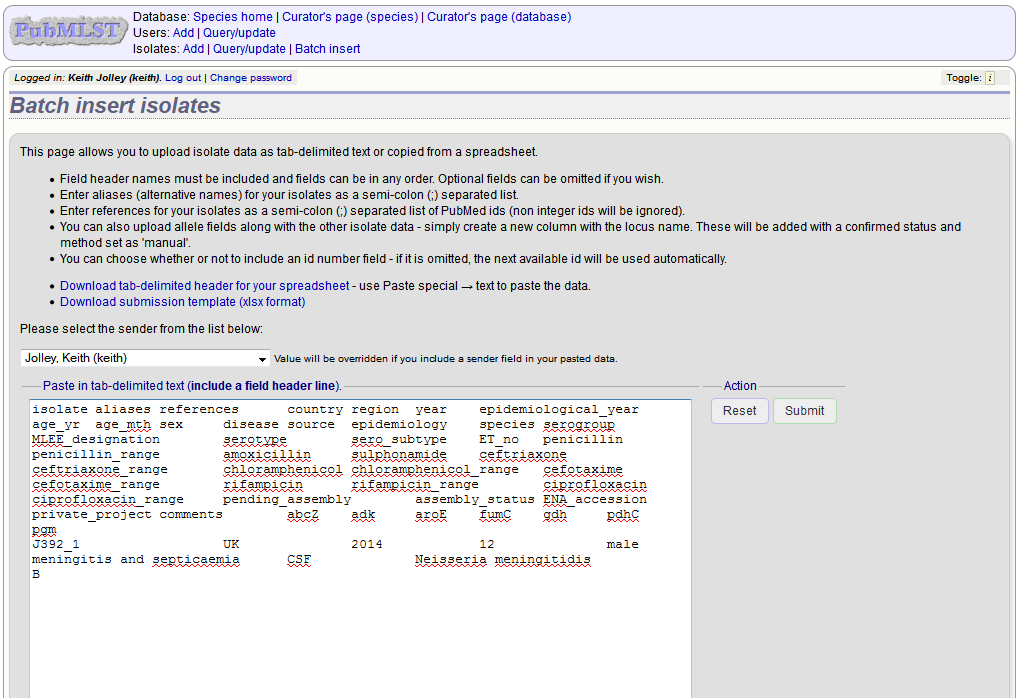
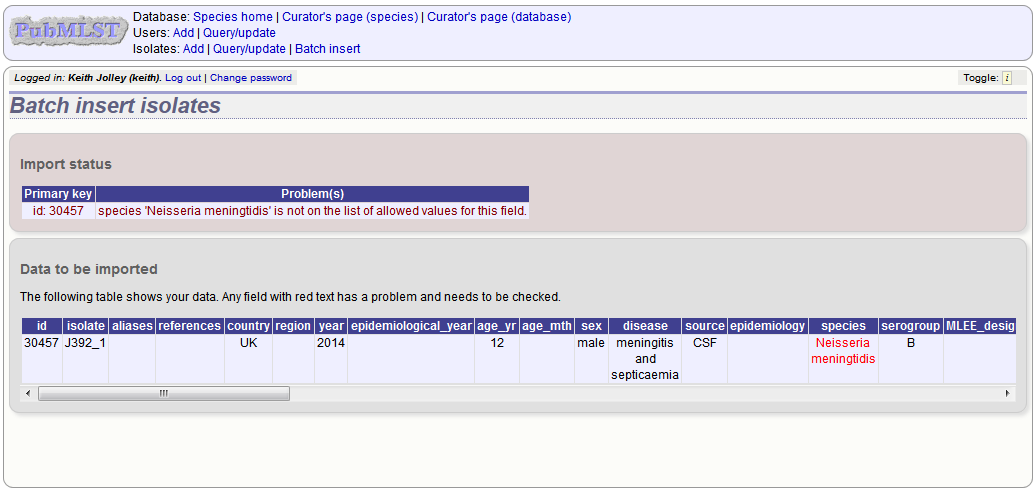
Press submit. Data are checked for consistency and if there are no problems you can then confirm the submission.
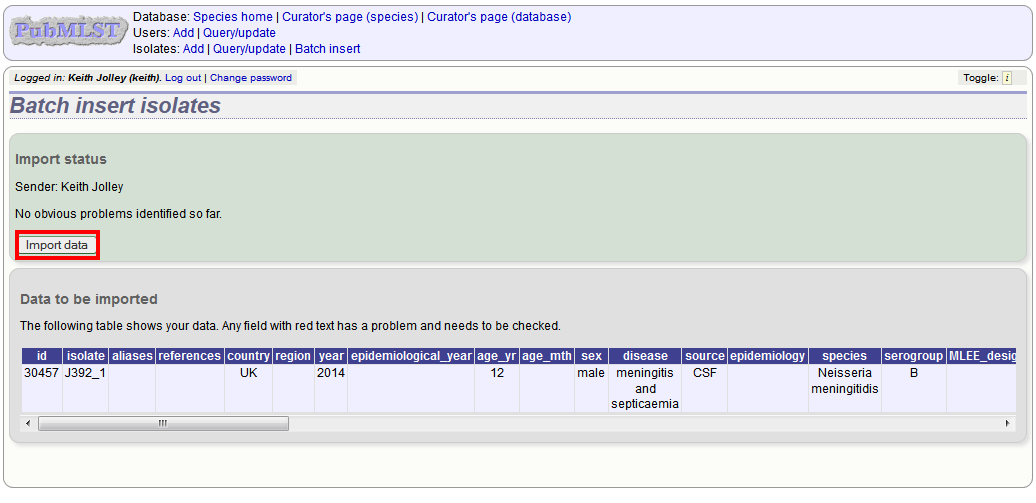
Any problems with the data will be listed and highlighted within the table. Fix the data and resubmit if this happens.
Updating and deleting single isolate records¶
First you need to locate the isolate record. You can either browse or use a search or list query.
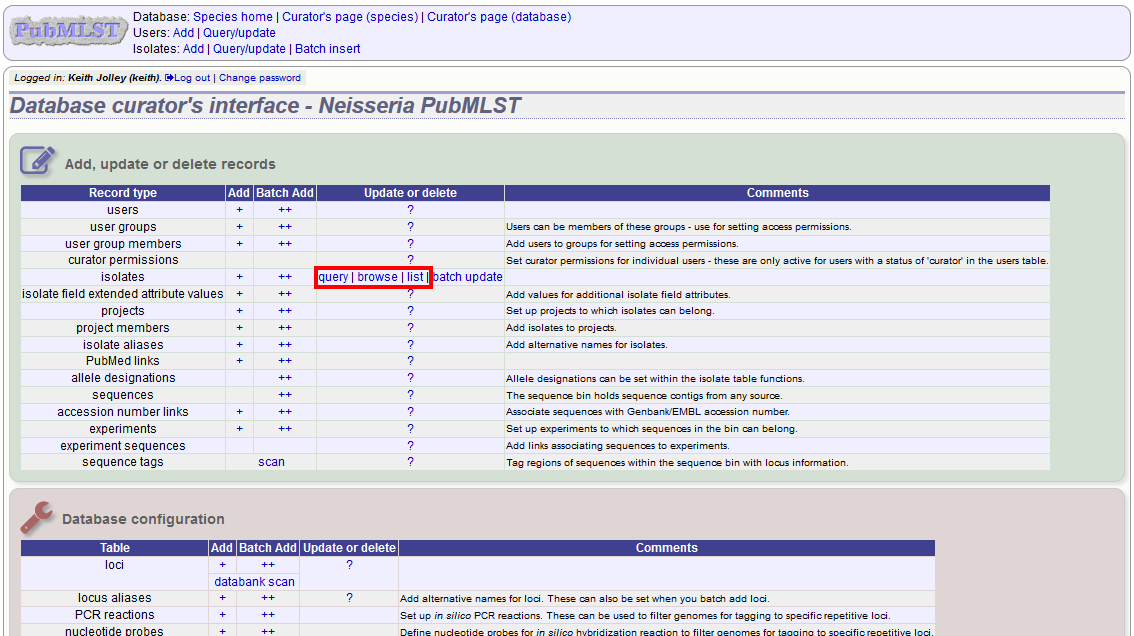
The query interface is the same as the public query interface. Following a query, a results table of isolates will be displayed. There will be delete and update links for each record.
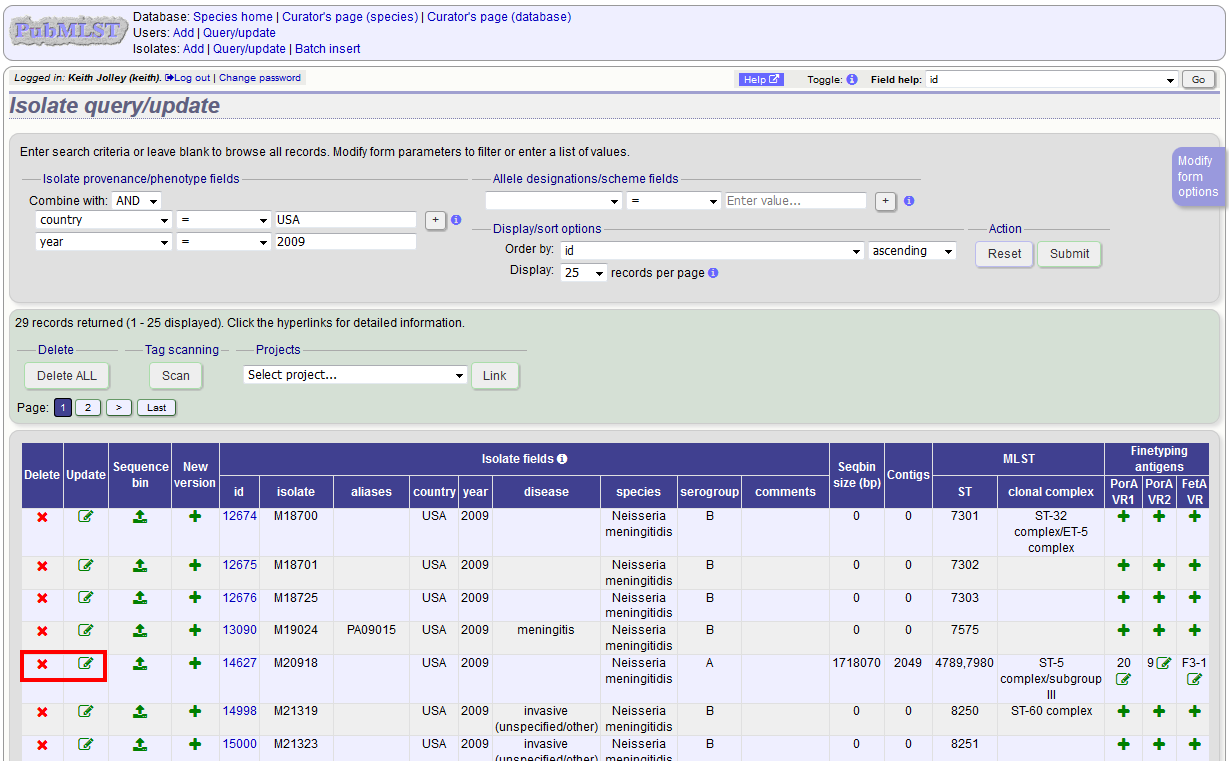
Clicking the ‘Delete’ link takes you to a page displaying the full isolate record.
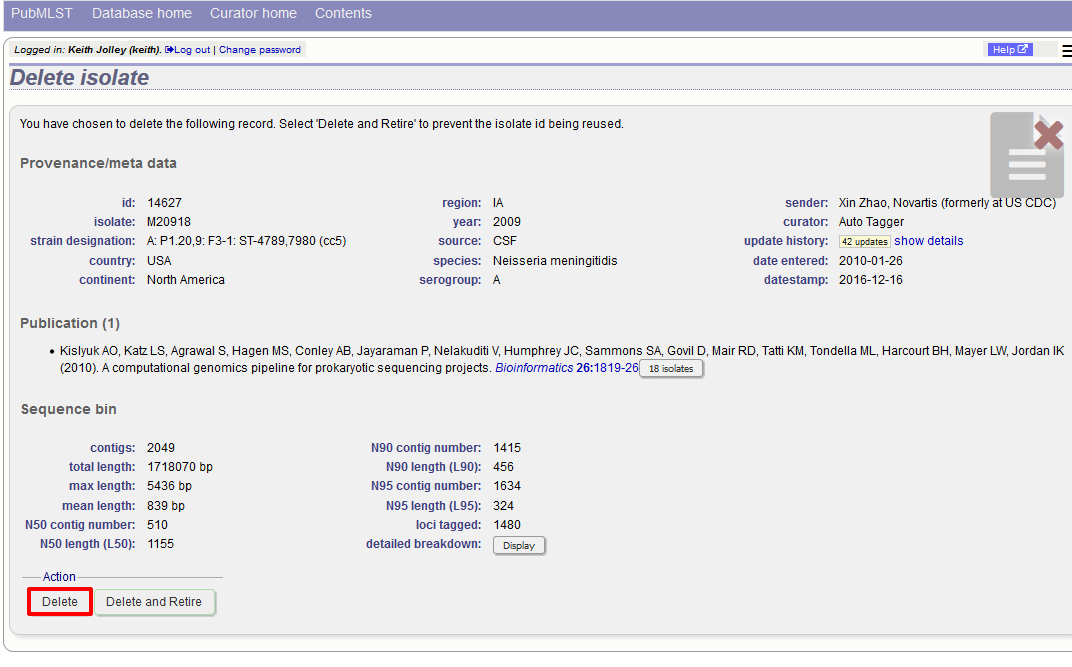
Pressing ‘Delete’ from this record page confirms the deletion.
Clicking the ‘Update’ link for an isolate takes you to an update form. Make the required changes and click ‘Update’.
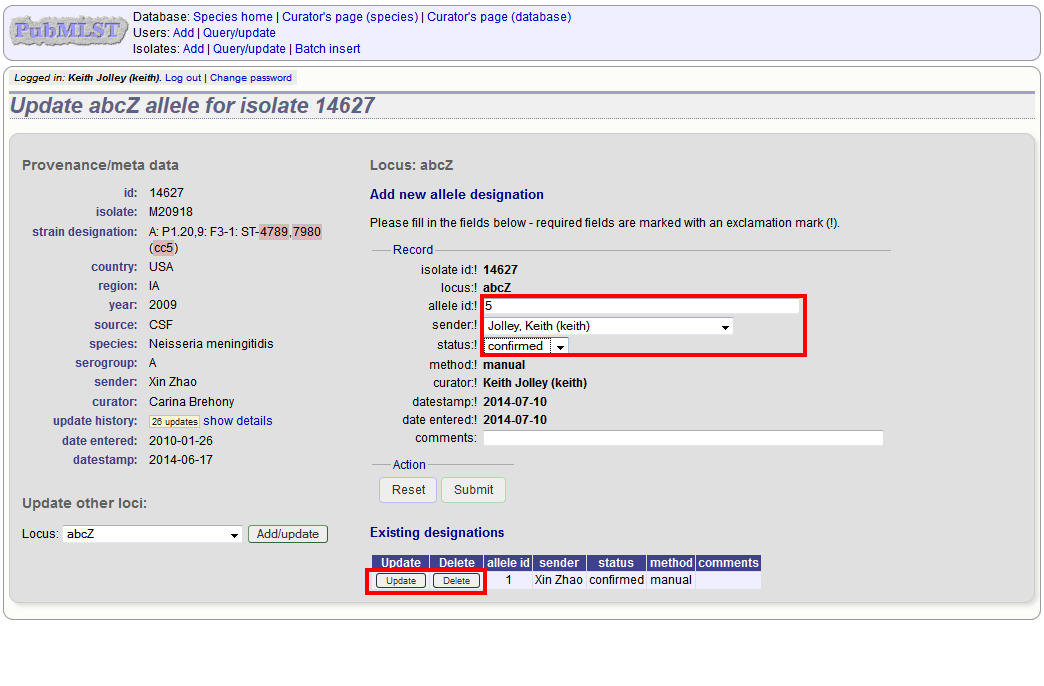
Allele designations can also be updated by clicking within the scheme tree and selecting the ‘Add’ or ‘Update’ link next to a displayed locus.
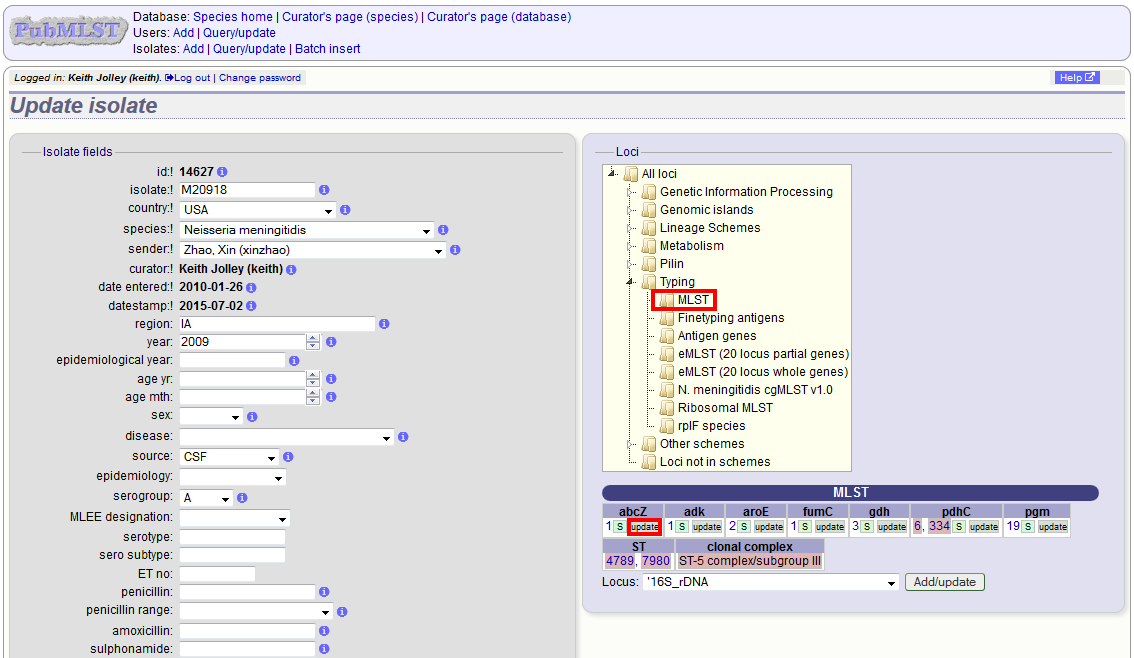
Schemes will only appear in the tree if data for at least one of the loci within the scheme has been added. You can additionally add or update allelic designations for a locus by choosing a locus in the drop-down list box and clicking ‘Add/update’.
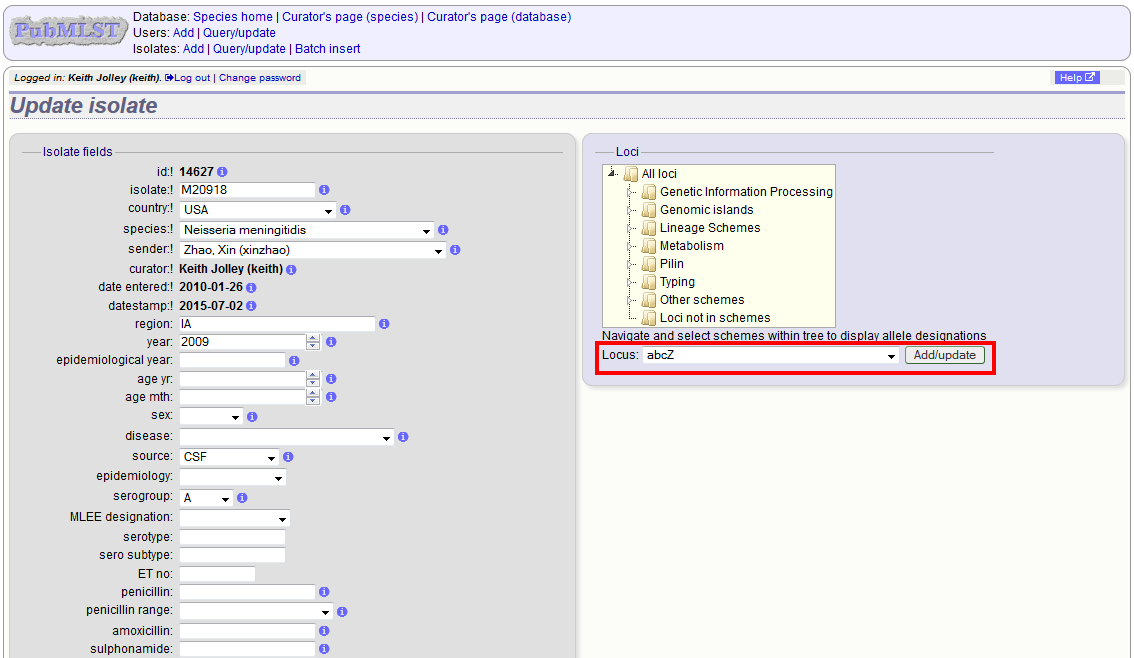
The allele designation update page allows you to modify an existing designation, or alternatively add additional designations. The sender, status (confirmed/provisional) and method (manual/automatic) needs to be set for each designation (all pending designations have a provisional status). The method is used to differentiate designations that have been determined manually from those determined by an automated algorithm.
Batch updating multiple isolate records¶
Select ‘batch update’ isolates link on the curator’s index page.
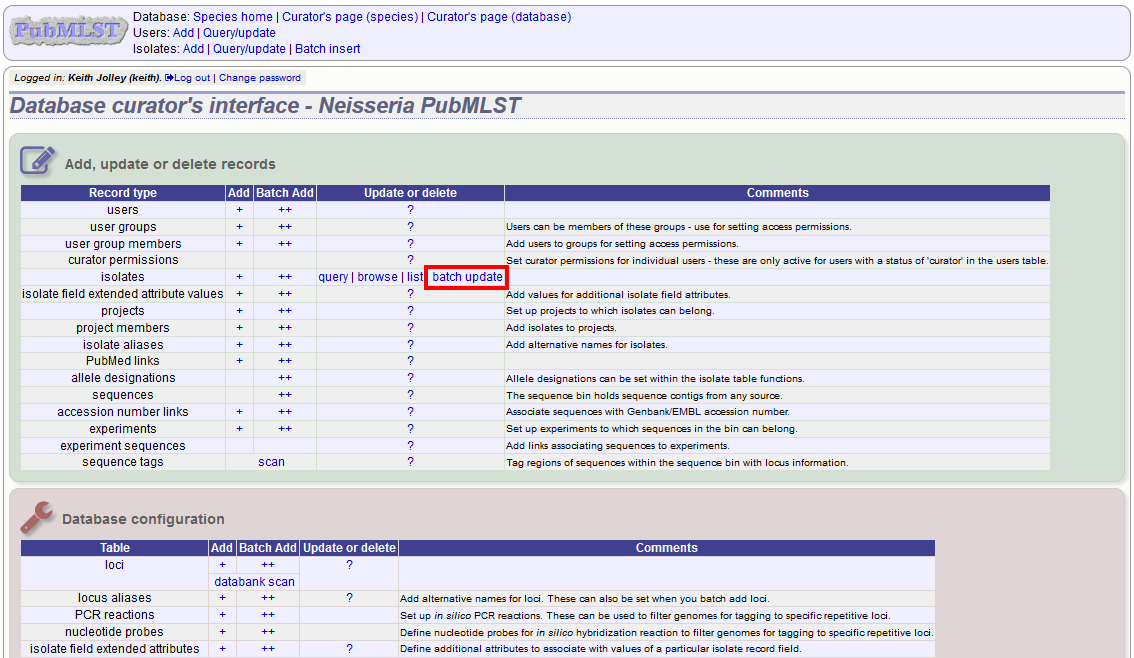
Prepare your update data in 3 columns in a spreadsheet:
- Unique identifier field
- Field to be updated
- New value for field
You should also include a header line at the top - this isn’t used so can contain anything but it should be present.
Columns must be tab-delimited which they will be if you copy and paste directly from the spreadsheet.
So, to update isolate id-100 and id-101 to serogroup B you would prepare the following:
id field value
100 serogroup B
101 serogroup B
Select the field you are using as a unique identifier, in this case id, from the drop-down list box, and paste in the data. If the fields already have values set, you should also check the ‘Update existing values’ checkbox. Press ‘submit’.
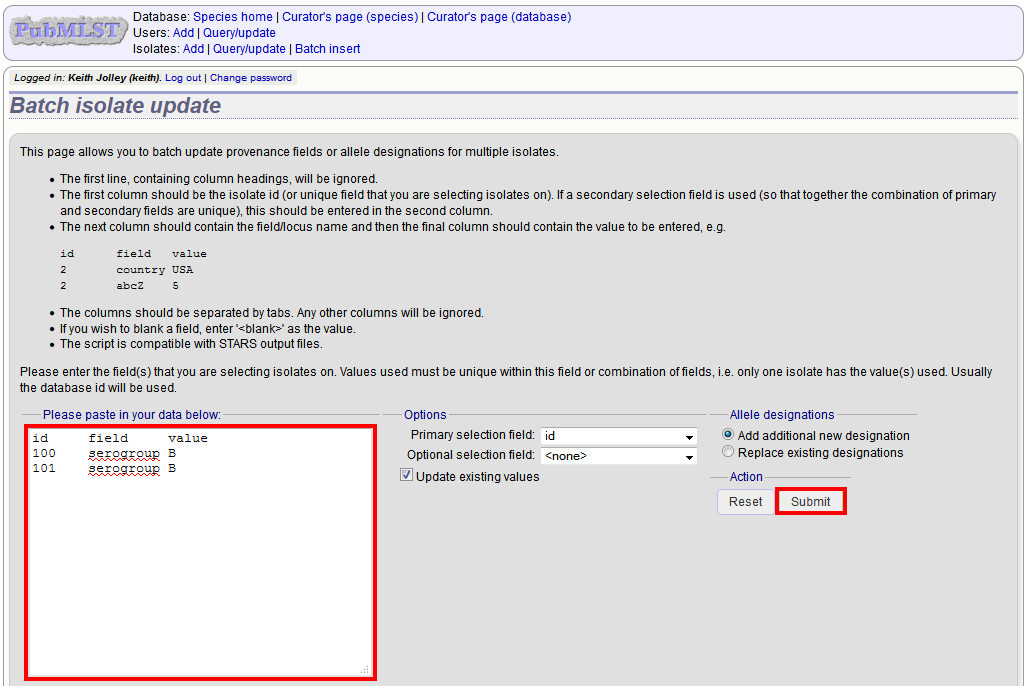
A confirmation page will be displayed if there are no problems. If there are problems, these will be listed. Press ‘Upload’ to upload the changes.
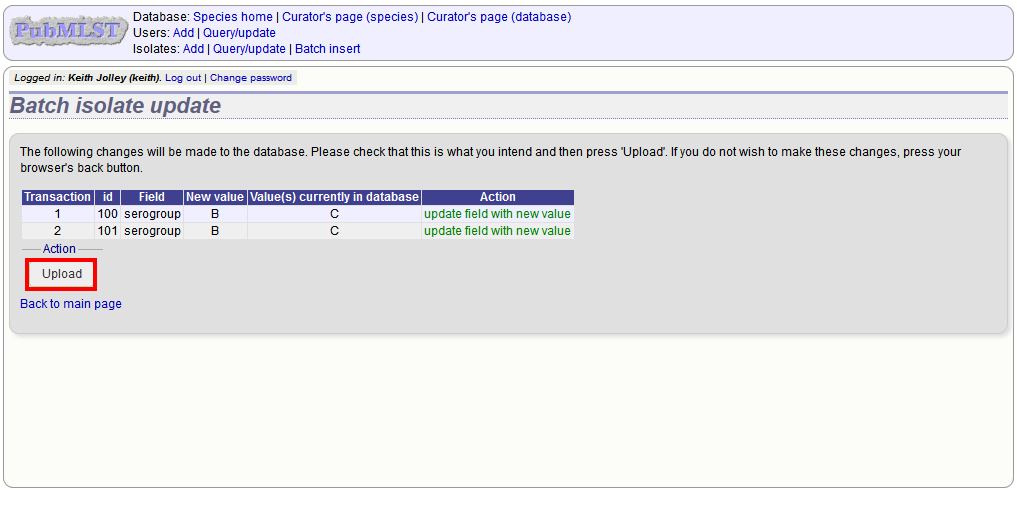
You can also use a secondary selection field such that a combination of two fields uniquely defines the isolate, for example using country and isolate name.
So, for example, to update the serogroups of isolates CN100 and CN103, both from the UK, select the appropriate primary and secondary fields and prepare the data as follows:
isolate country field value
CN100 UK serogroup B
CN103 UK serogroup B
Deleting multiple isolate records¶
Note
Please note that standard curator accounts may not have permission to delete multiple isolates. Administrator accounts are always able to do this.
Before you can delete multiple records, you need to search for them. From the curator’s main page, click the Query isolates link:
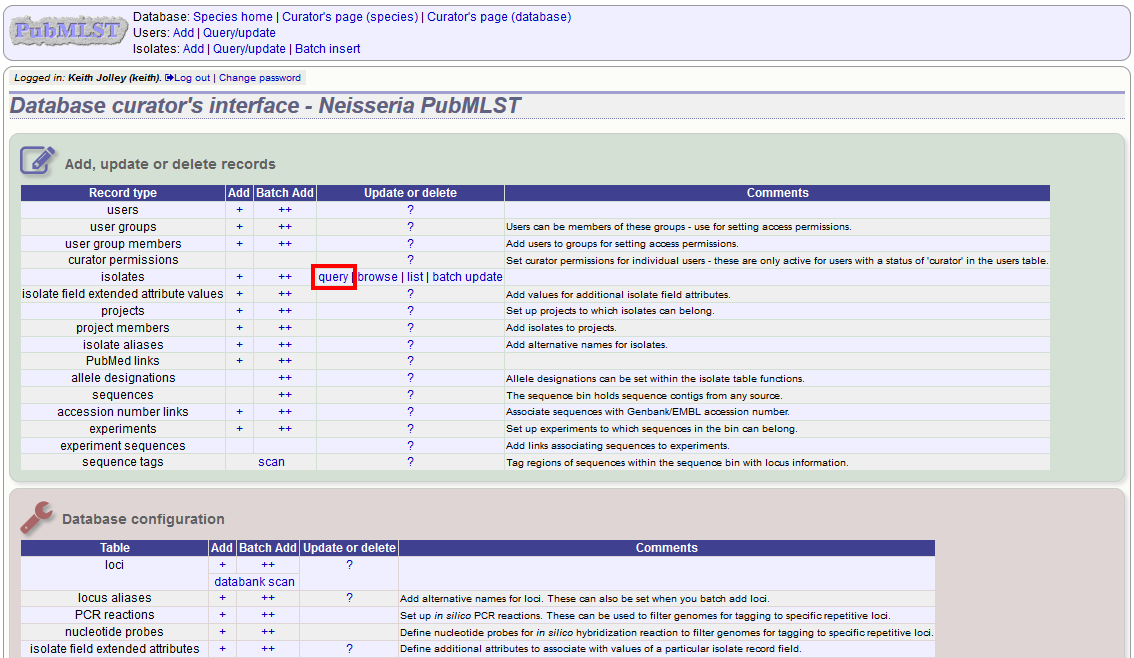
Enter search criteria that specifically return the isolates you wish to delete. Click ‘Delete ALL’.
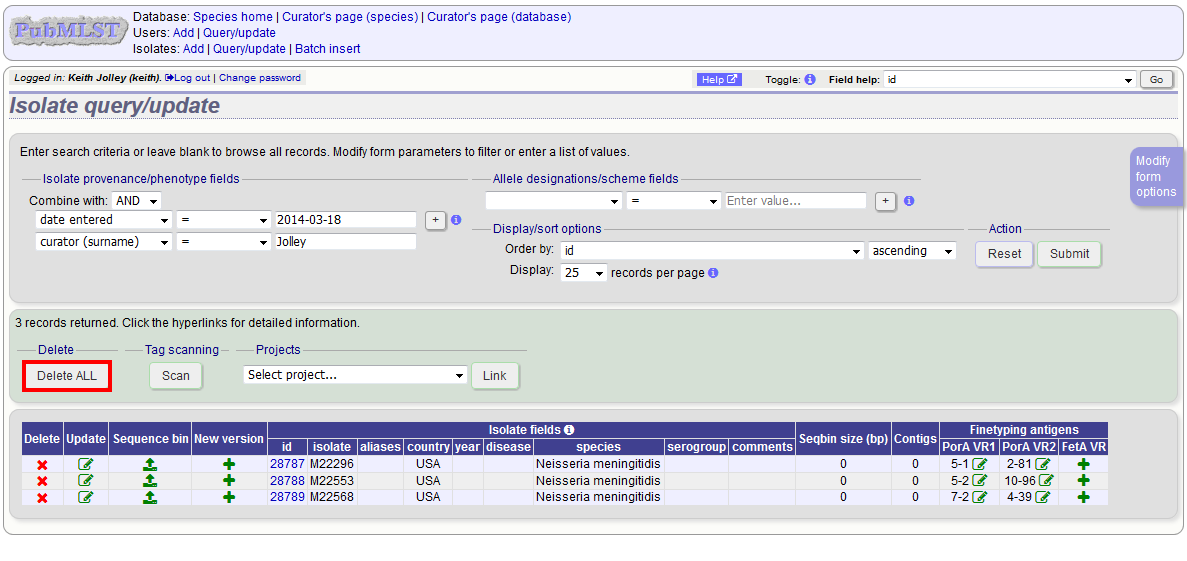
You will have a final chance to change your mind:
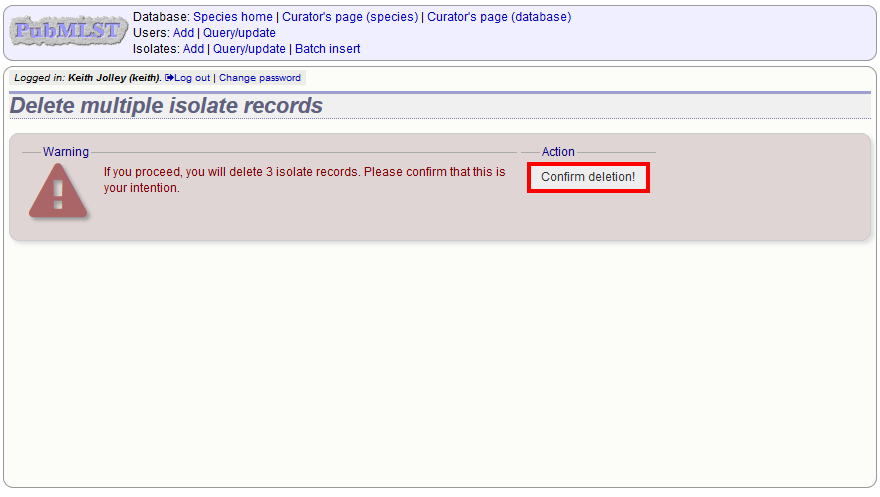
Click ‘Confirm deletion!’.
Retiring isolate identifiers¶
Sometimes there is a requirement to prevent the automated assignment of a particular isolate identifier number - an isolate with that identifier may have been commonly used and has since been removed. Reassignment of the identifier to a new isolate record may lead to confusion, so in this instance, it would be better to prevent this.
You can retire an isolate identifier by clicking the ‘Add’ retired isolates link on the isolates database curators’ page.
Enter the isolate id to retire and click ‘Submit’.
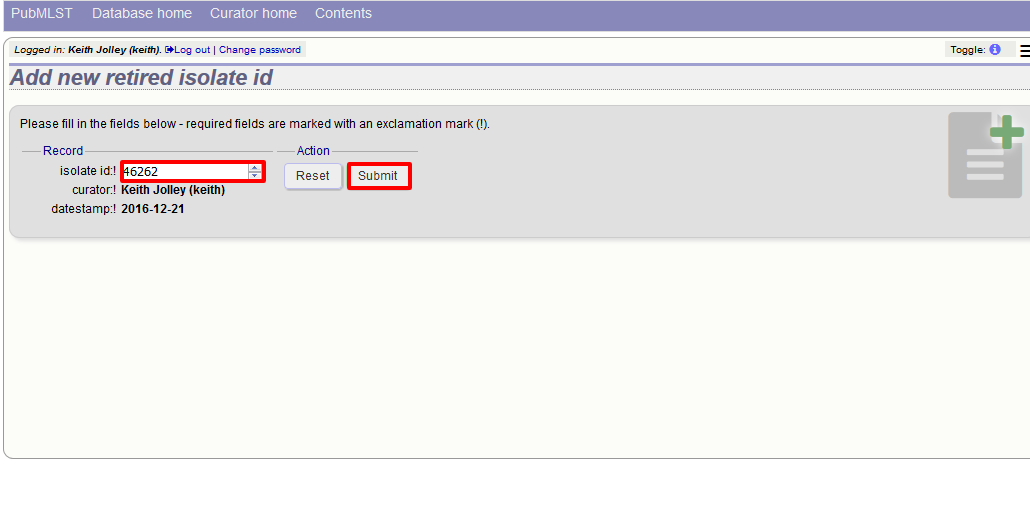
You cannot retire an isolate identifier that already exists, so you must delete it before retiring it. Once an identifier is retired, you will not be able to create a new isolate record using that identifier.
You can also retire an isolate identifier when you delete an isolate record.
Setting alternative names for isolates (aliases)¶
Isolates can have any number of alternative names that they are known by. These isolate aliases can be set when isolates are first added to the database or batch uploaded later. When querying by isolate names, the aliases are also searched automatically.
If adding isolates singly, add the aliases in to the aliases box (one alias per line):
If batch adding isolates, they can be entered as a semi-colon (;) separated list in the aliases column.
As stated above, aliases can also be batch added. To do this, click the batch add (++) link on the curator’s index page.
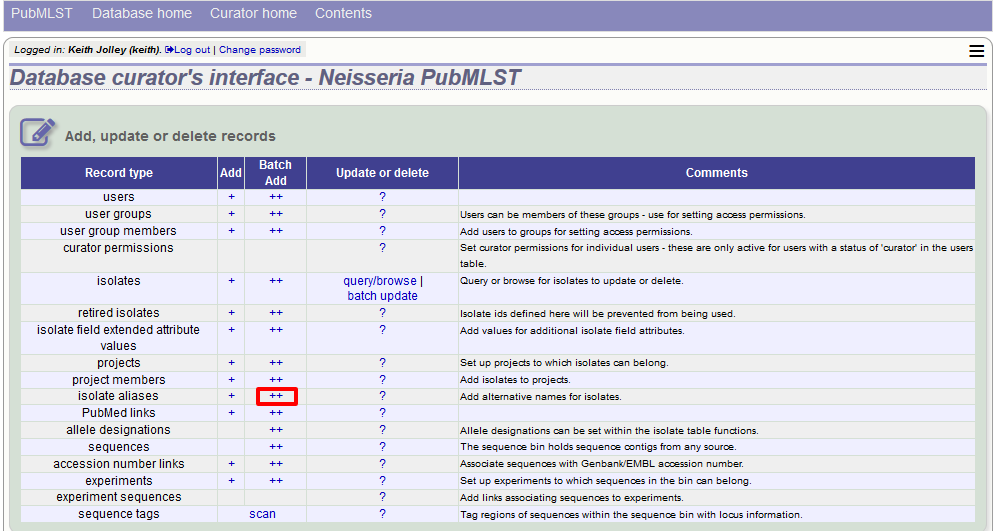
Prepare a list in a spreadsheet using the provided template. This consists of two columns: isolate_id and alias. For example, to add the aliases ‘JHS212’ and ‘NM11’ to isolate id 5473, the values to paste in look like:
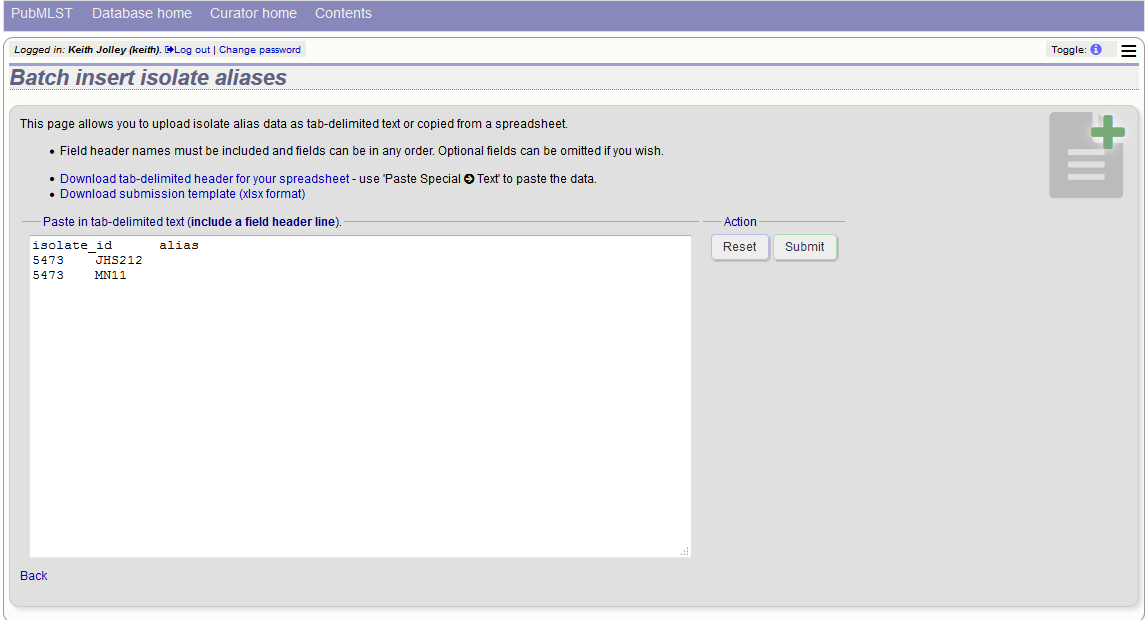
A confirmation page will be displayed.
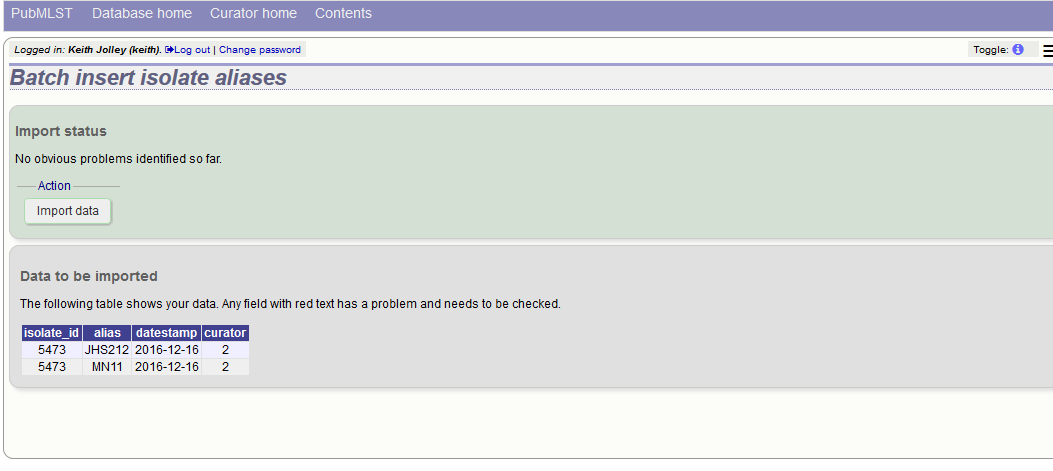
Click ‘import data’.
Linking isolate records to publications¶
Isolates can be associated with publications by adding PubMed id(s) to the record. This can be done when adding the isolate, where lists of PubMed ids can be entered in to the web form.
They can also be associated in batch after the upload of isolate records. Click the PubMed links batch add (++) link on the curator’s main page.
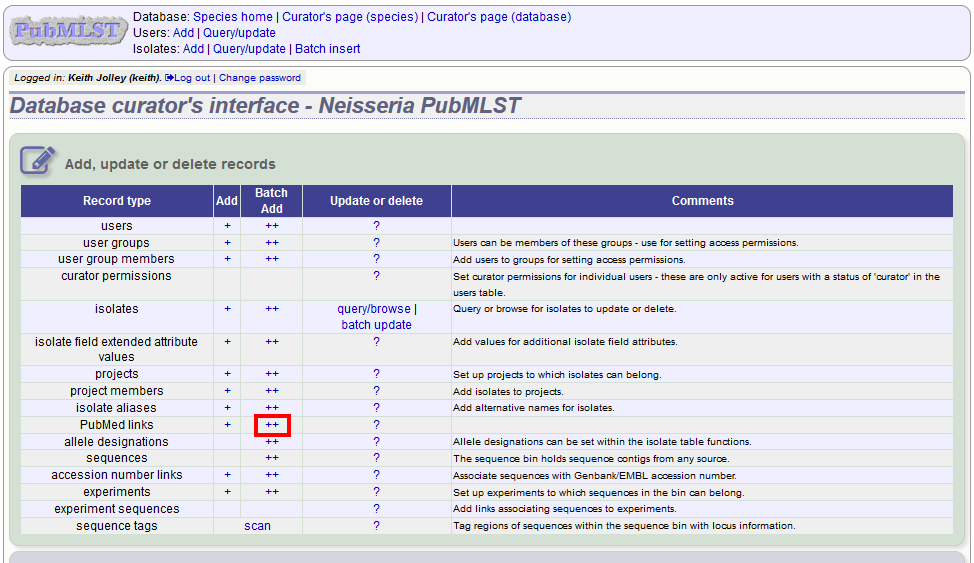
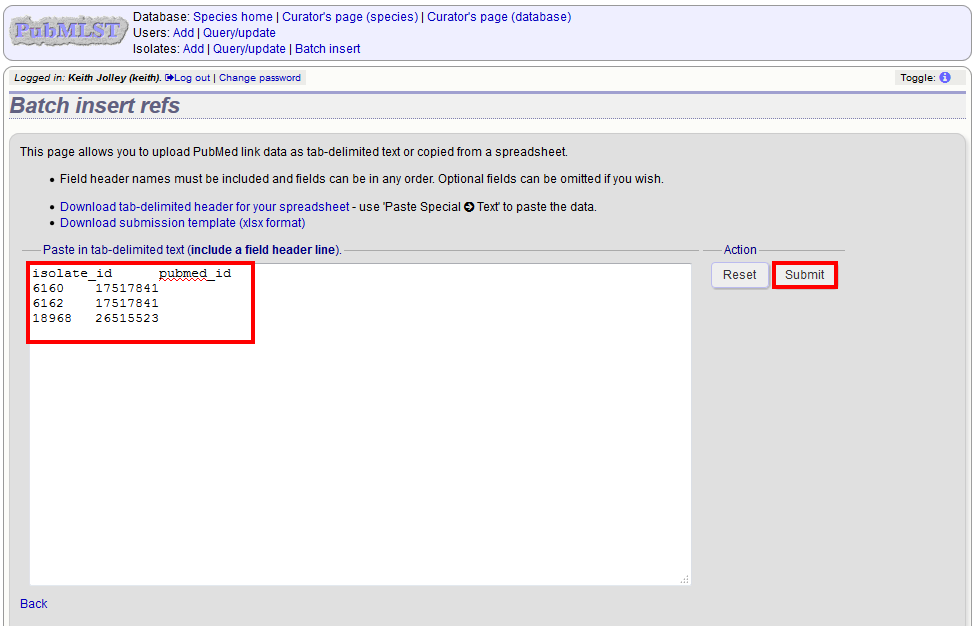
Open the Excel template by clicking the link.

The Excel template has two columns, isolate_id and pubmed_id. Simply fill this in with a line for each record and then paste the entire spreadsheet in to the web form and press submit.
To ensure that publication information is stored locally and available for searching, the references database needs to be updated regularly.
Uploading sequence contigs linked to isolate records¶
Select isolate from drop-down list¶
To upload sequence data, click the sequences batch add (++) link on the curator’s main page.
Select the isolate that you wish to link the sequence to from the dropdown list box. You also need to enter the person who sent the data. Optionally, you can add the sequencing method used.
Paste sequence contigs in FASTA format in to the form.
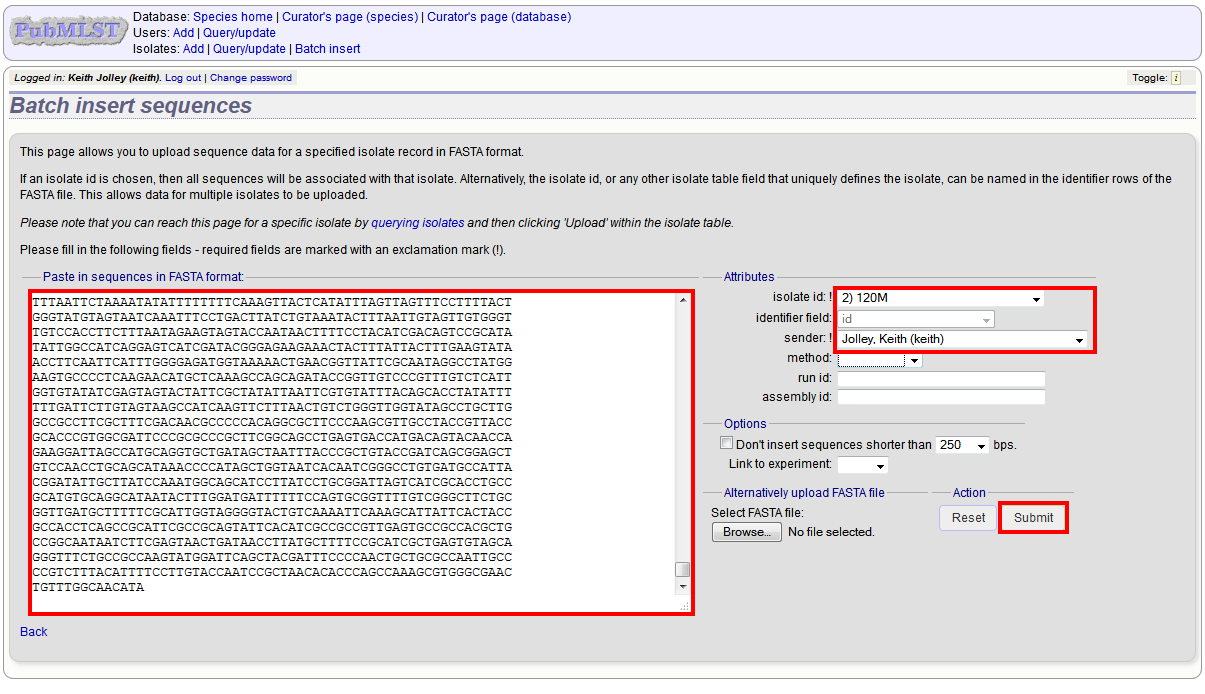
Click ‘Submit’. A summary of the number of isolates and their lengths will be displayed. To confirm upload, click ‘Upload’.
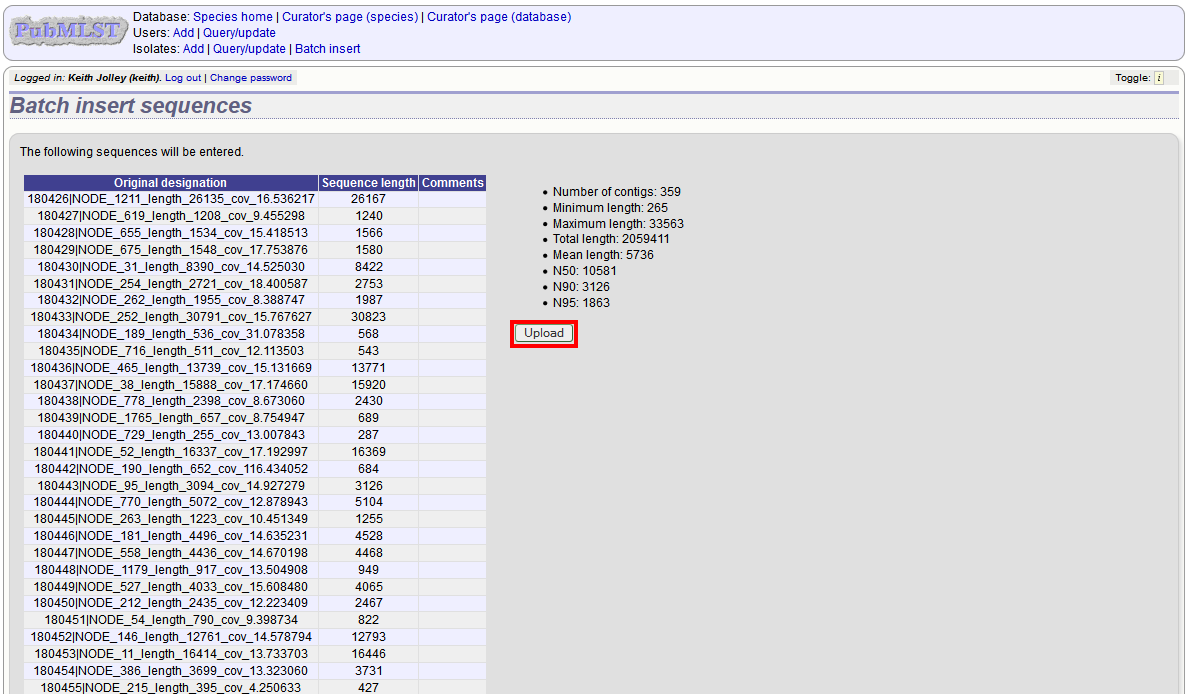
It is also possible to upload data for multiple isolates at the same time, but these must exist as single contigs for each isolate. To do this, select ‘Read identifier from FASTA’ in the isolate id field and select the field that you wish to use as the identifier in the ‘identifier field’, e.g. to use isolate names select ‘isolate’ here.
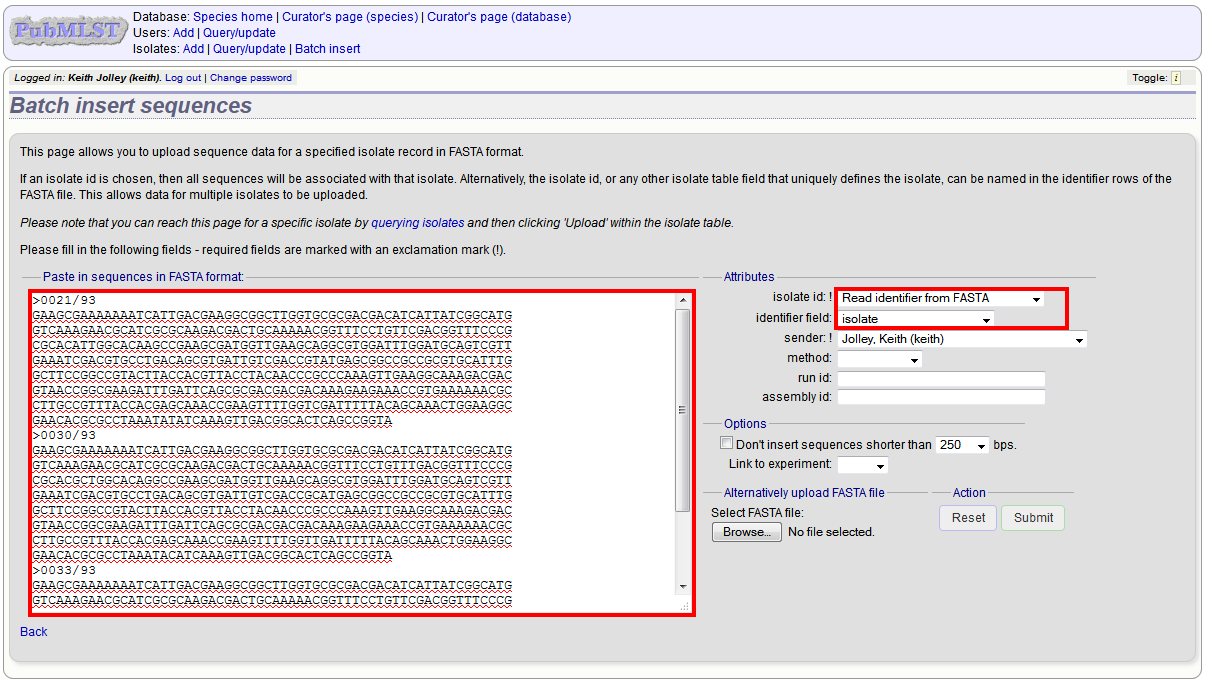
Provided the identifier used uniquely identifies the isolate you will get a confirmation screen. If the isolate name does not do this you’ll probably have to use the database id number instead. Click ‘Upload’ to confirm.
Select from isolate query¶
As an alternative to selecting the isolate from a dropdown list (which can become unwieldy for large databases), it is also possible to upload sequence data following an isolate query.
Click the isolate query link from the curator’s main page.
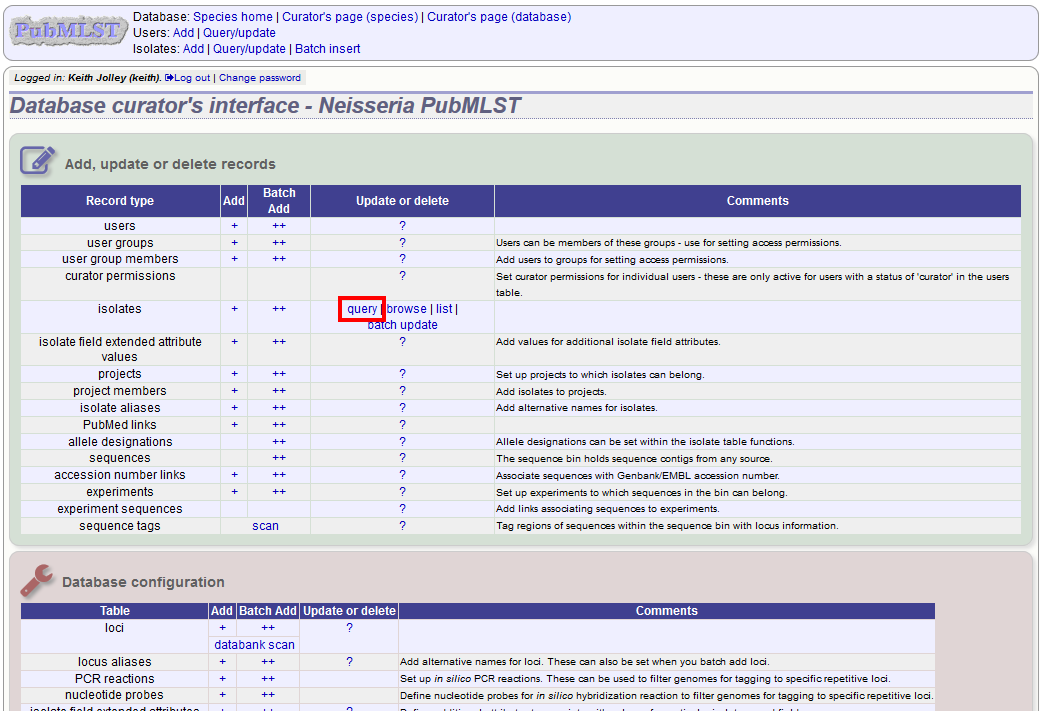
Enter your search criteria. From the list of isolates displayed, click the ‘Upload’ link in the sequence bin column of the appropriate isolate record.
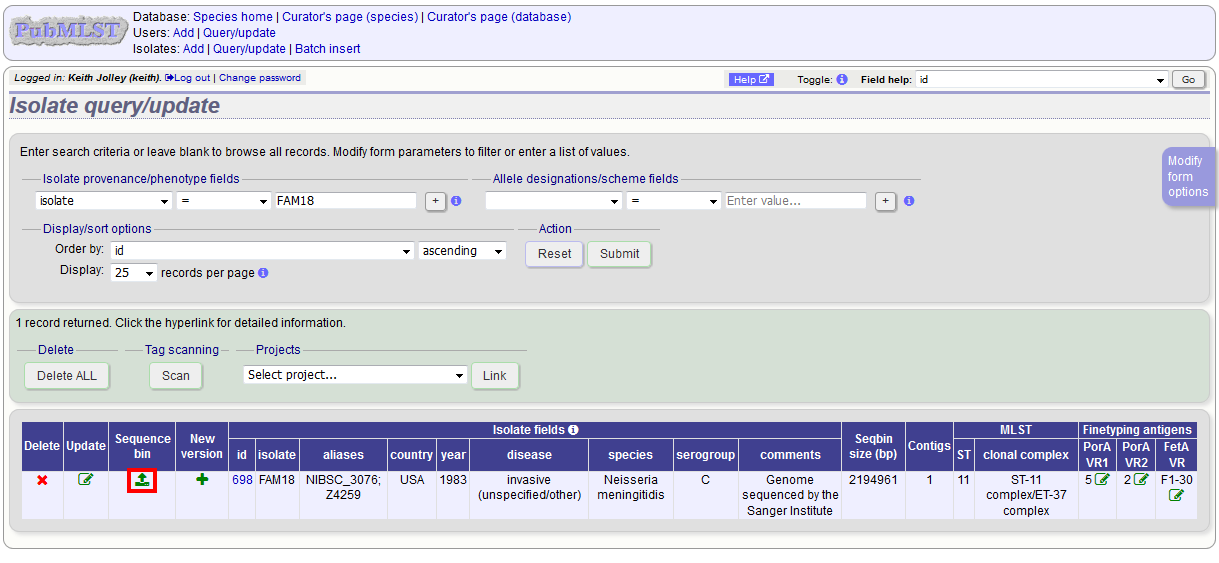
The same upload form as detailed above is shown. Instead of a dropdown list for isolate selection, however, the chosen isolate will be pre-selected.
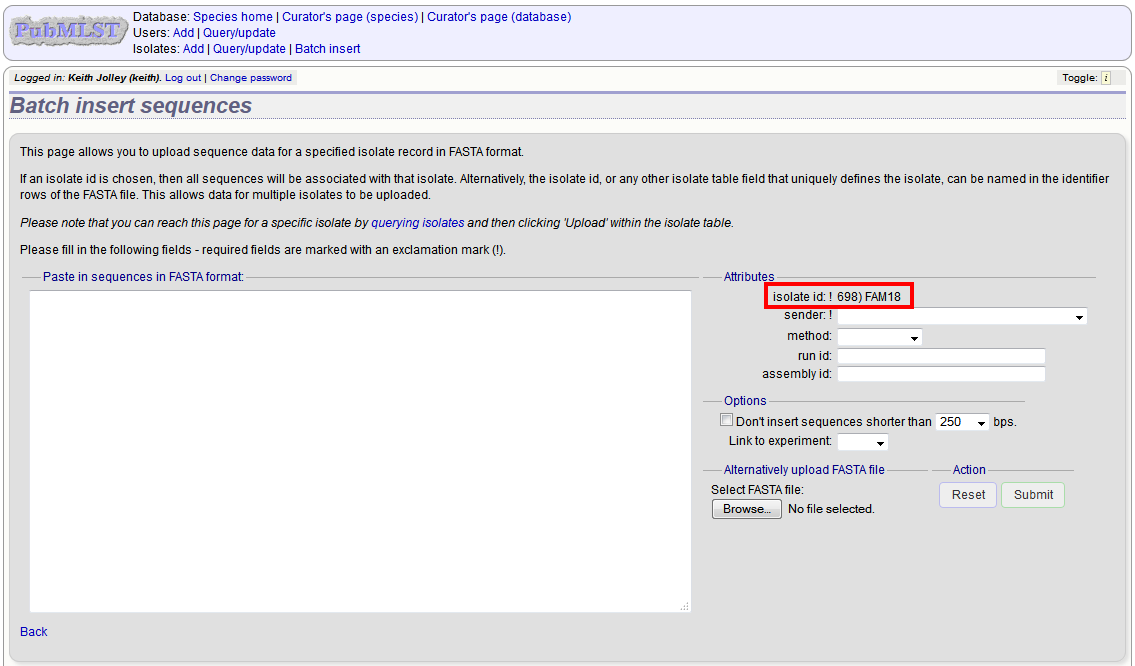
Upload options¶
On the upload form, you can select to filter out short sequences from your contig list.
If your database has experiments defined (experiments are used for grouping sequences and can be used to filter the sequences used in tag scanning), you can also choose to upload your contigs as part of an experiment. To do this, select the experiment from the dropdown list box.
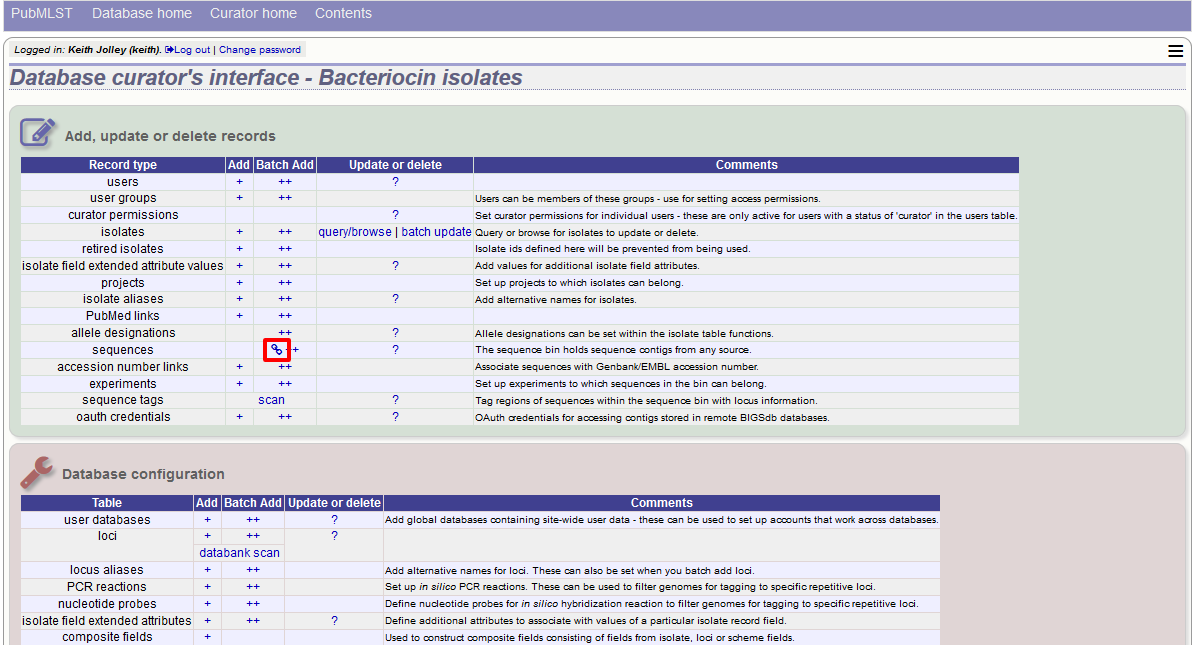
Linking remote contigs to isolate records¶
If remote contigs have been enabled, isolates can be linked to contigs stored in an external BIGSdb database, rather than directly uploaded. These well then be loaded when needed, for example during scanning or data export. This will be marginally slower than hosting contigs within the same database, but minimises duplication of sequence data and associated storage. Contigs need to be accessible via the BIGSdb RESTful API.
Click the sequences link icon on the curator’s main page.
Either select the isolate id from the dropdown list, or enter it manually (list is disabled if there are >1000 records in the database). Enter the URI for the RESTful API of the parent isolate record, e.g. http://rest.pubmlst.org/db/pubmlst_rmlst_isolates/isolates/933. This URI can require authentication if credentials have been set up.
Press submit.
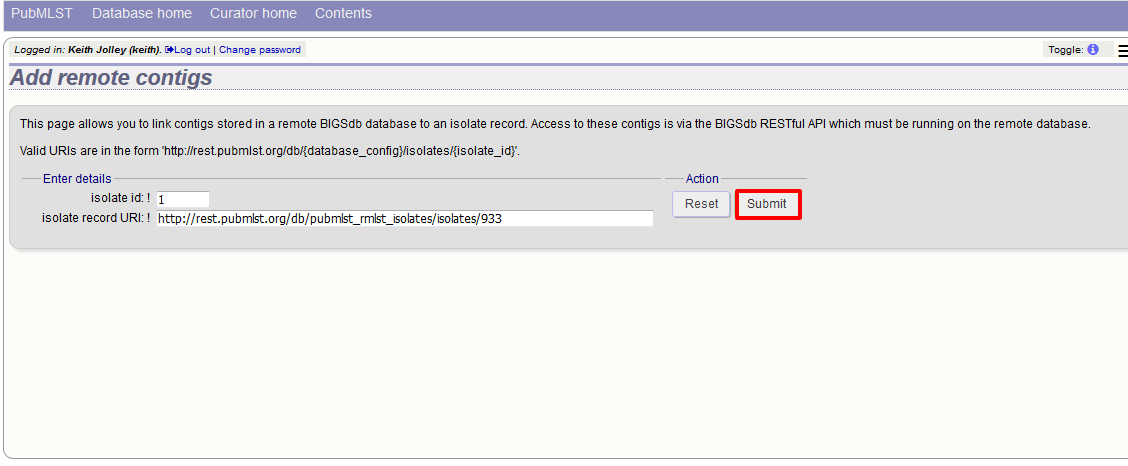
Summary information about the number of contigs and their total length will be downloaded from the remote isolate record. You will then be prompted to upload this information to the database, by clicking the ‘Upload’ button.
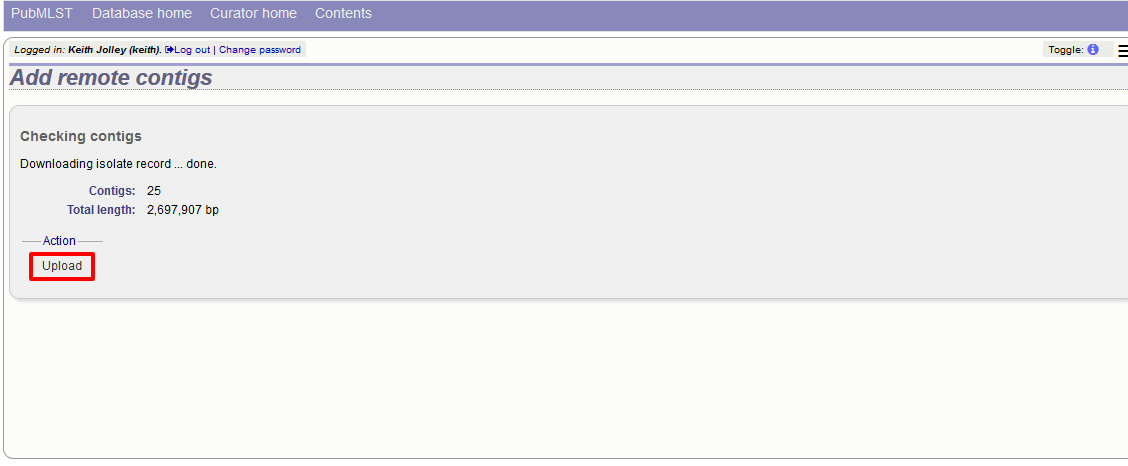
The contigs will be downloaded in bulk in order to determine their lengths. This information is stored within the local database as it is required for various outputs. Full metadata is not stored at this stage.
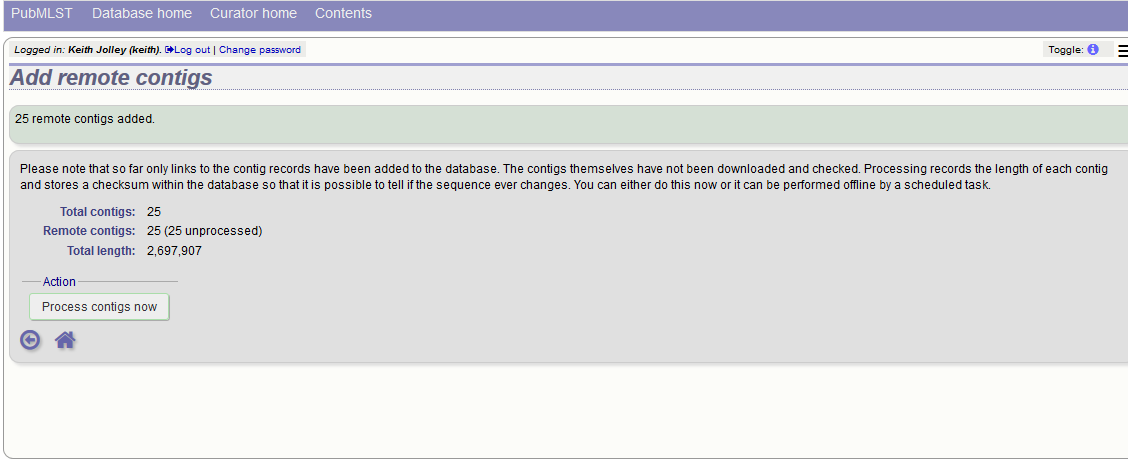
This is all that is required for the contigs to be used as normal. In order to get the full metadata about the contigs (sequencing platform used, sender and datestamp information), you can choose to process the contigs by clicking the ‘Process contigs now’ button. This will download each contig in turn, and store its provenance metadata locally.
Alternatively, this step can be performed offline automatically.
Automated web-based sequence tagging¶
Sequence tagging, or tag-scanning, is the process of identifying alleles by scanning the sequence bin linked to an isolate record. Defined loci can either have a single reference sequence, that is defined in the locus table, or they can be linked to an external database that contains the sequences for known alleles. The tagging function uses BLAST to identify sequences and will tag the specific sequence region with locus information and an allele designation if a matching allele is identified by reference to an external database.
Select ‘scan’ sequence tags on the curator’s index page.
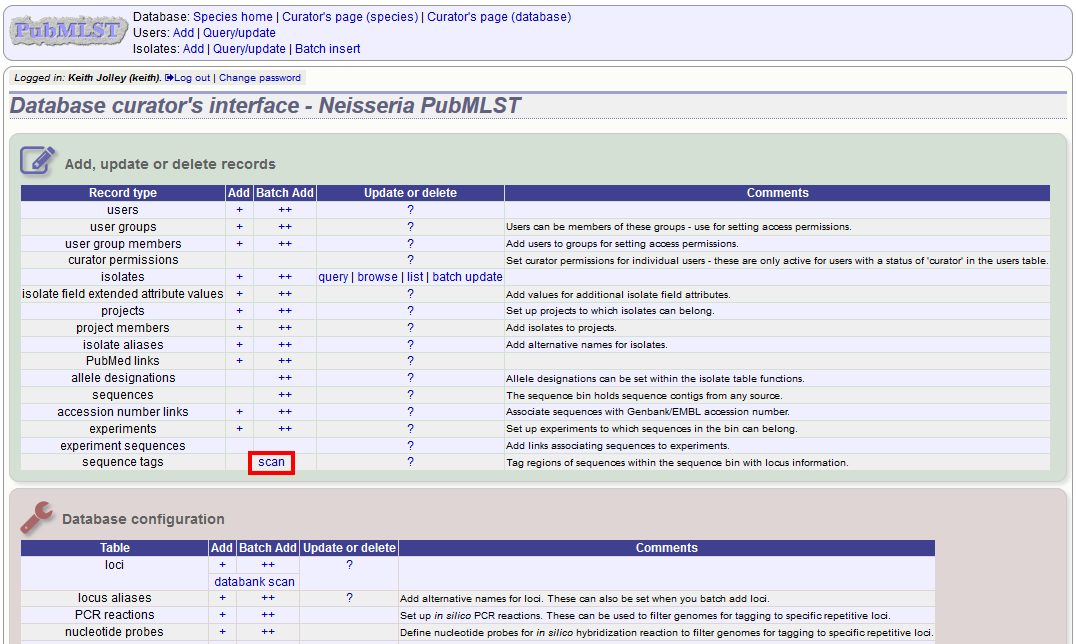
Next, select the isolates whose sequences you wish to scan against. Multiple isolates can be selected by holding down the Ctrl key. All isolates can be selected by clicking the ‘All’ button under the isolate selection list.
Select either individual loci or schemes (collections of loci) to scan against. Again, multiple selections can be made.
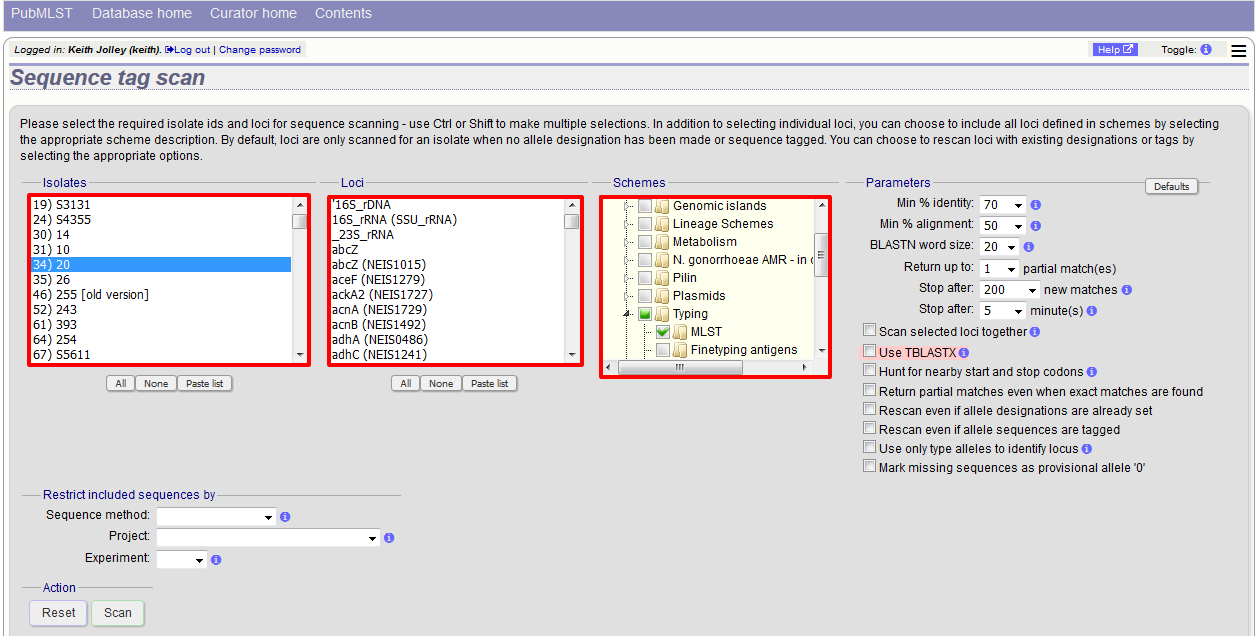
Choose your scan parameters. Lowering the value for BLASTN word size will increase the sensitivity of the search at the expense of time. Using TBLASTX is more sensitive but also much slower. TBLASTX can only be used to identify the sequence region rather than a specific allele (since it will only match the translated sequence and there may be multiple alleles that encode a particular peptide sequence).
By default, for each isolate only loci that have not had either an allele designation made or a sequence region scanned will be scanned again. To rescan in these cases, select either or both the following:
- Rescan even if allele designations are already set
- Rescan even if allele sequences are tagged
You can select to only use type alleles to identify the locus. This will constrain the search space so that allele definitions don’t become more variable over time. If a partial match is found to a type allele then a full database lookup will be performed to identify any known alleles. An allele can be given a status of type allele when defining.
If fast scanning is enabled, there will also be an option to ‘Scan selected loci together’. This can be significantly quicker than a locus-by-locus search against all alleles but is not enabled by default as it can use more memory on the server and requires exemplar alleles to be defined.
Options can be returned to their default setting by clicking the ‘Defaults’ button.
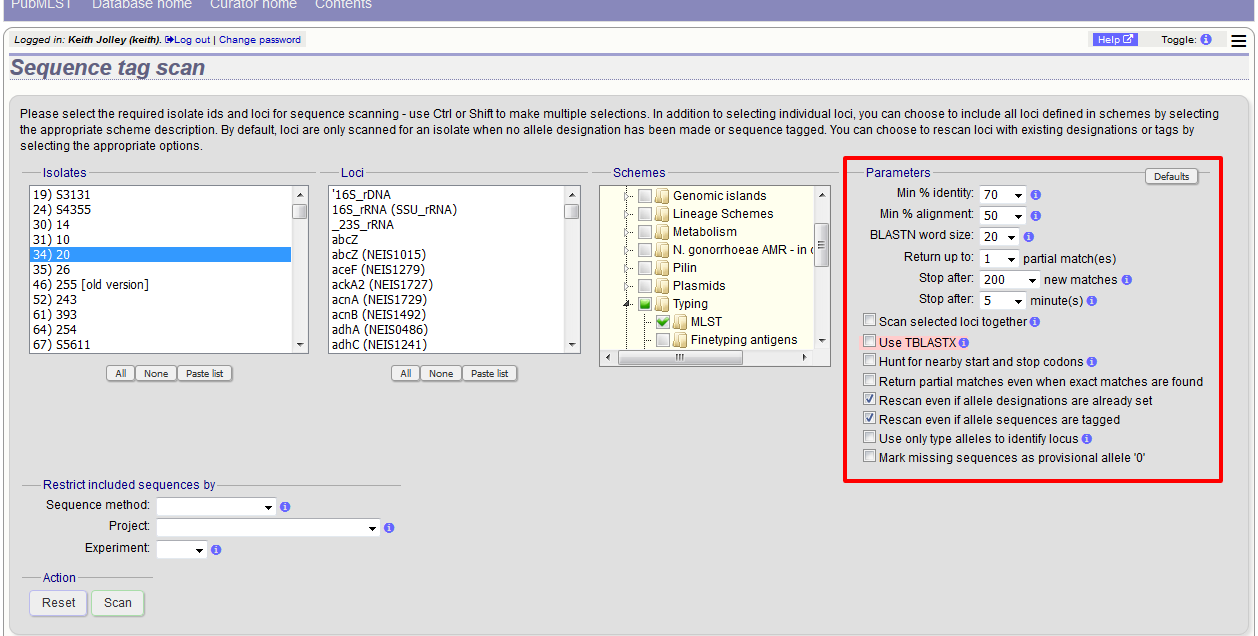
Press ‘Scan’. The system takes approximately 1-2 seconds to identify each sequence (depending on machine speed and size of definitions databases). Alternatively, if ‘Scan selected loci together’ is available and selected, it may take longer to return initial results but total time should be less (e.g. a 2000 loci cgMLST scheme may be returned in 1-2 minutes). Any identified sequences will be listed in a table, with checkboxes indicating whether allele sequences or sequence regions are to be tagged.
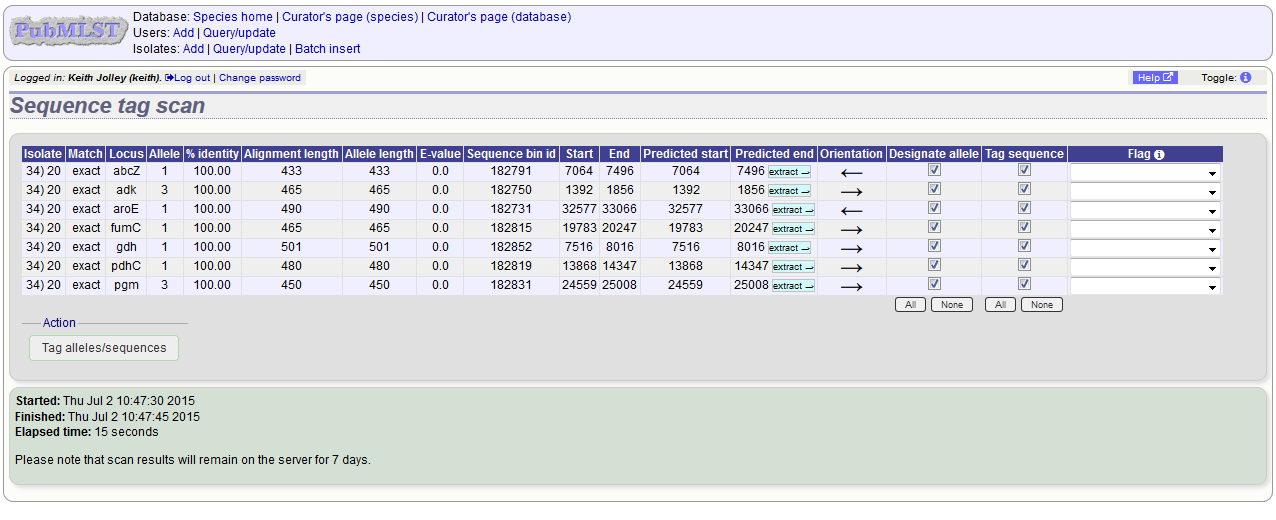
Individual sequences can be extracted for inspection by clicking the ‘extract →’ link. The sequence (along with flanking regions) will be opened in another browser window or tab.
Checkboxes are enabled against any new sequence region or allele designation. You can also set a flag for a particular sequence to mark an attribute. These will be set automatically if these have been defined within the sequence definition database for an identified allele.
See also
Ensure any sequences you want to tag are selected, then press ‘Tag alleles/sequences’.
If any new alleles are found, a link at the bottom will display these in a format suitable for automatic allele assignment by batch uploading to sequence definition database.
Projects¶
Creating the project¶
The first step in grouping by project is to set up a project.
Click the add (+) project link on the curator’s main page.
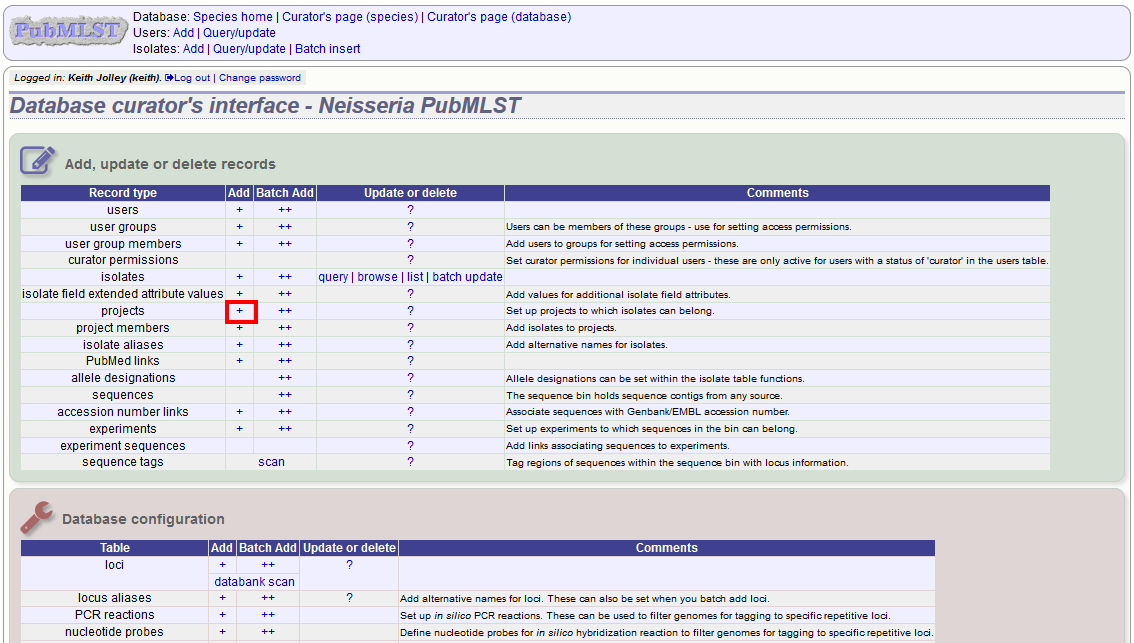
Enter a short description for the project. This is used in drop-down list boxes within the query interfaces, so make sure it is not too long.
You can also enter a full description. If this is added, the project description can displayed at the top of an isolate information page (but see ‘isolate_display’ flag below). The full description can include HTML formatting, including image links.
There are additionally two flags that affect how projects are listed:
- isolate_display - Setting this is required for the project and its description to be listed at the top of an isolate record (default: false).
- list - Setting this is required for the project to be listed in a page of projects linked from the main contents page.
Click ‘Submit’.
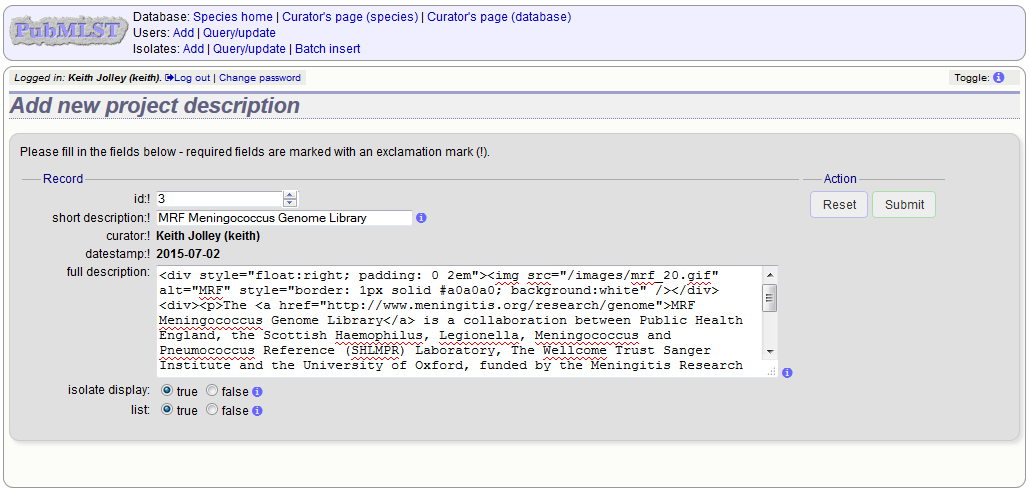
Explicitly adding isolates to a project¶
Explicitly adding isolates to the project can be done individually or in batch mode. To add individually, click the add (+) project member link on the curator’s main page.
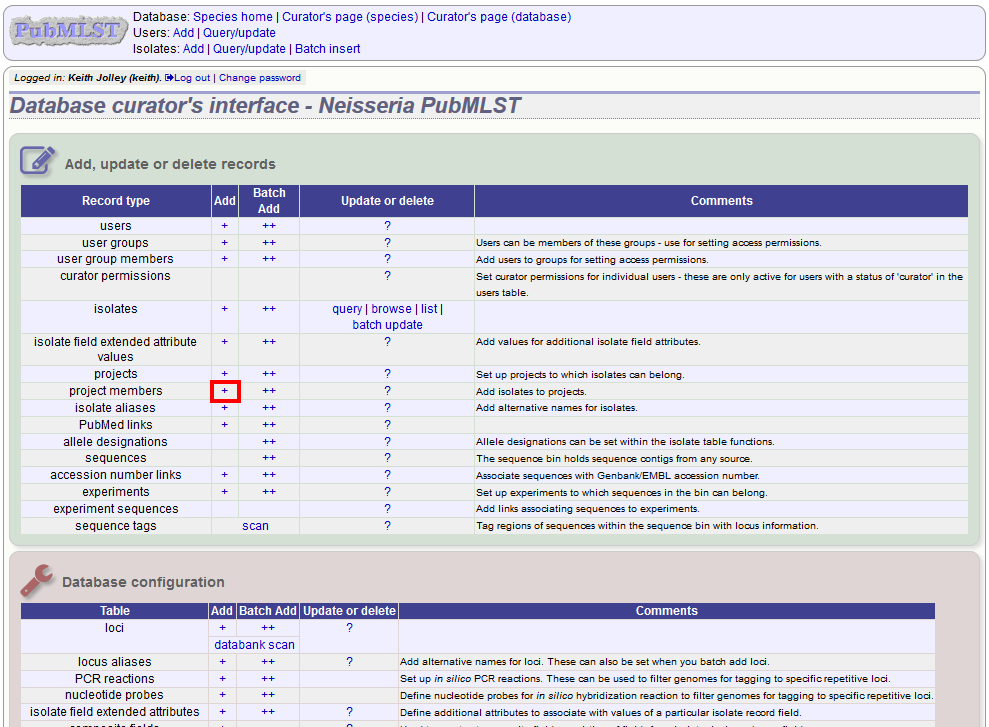
Select the project from the dropdown list box and enter the id of the isolate that you wish to add to the project. Click ‘Submit’.
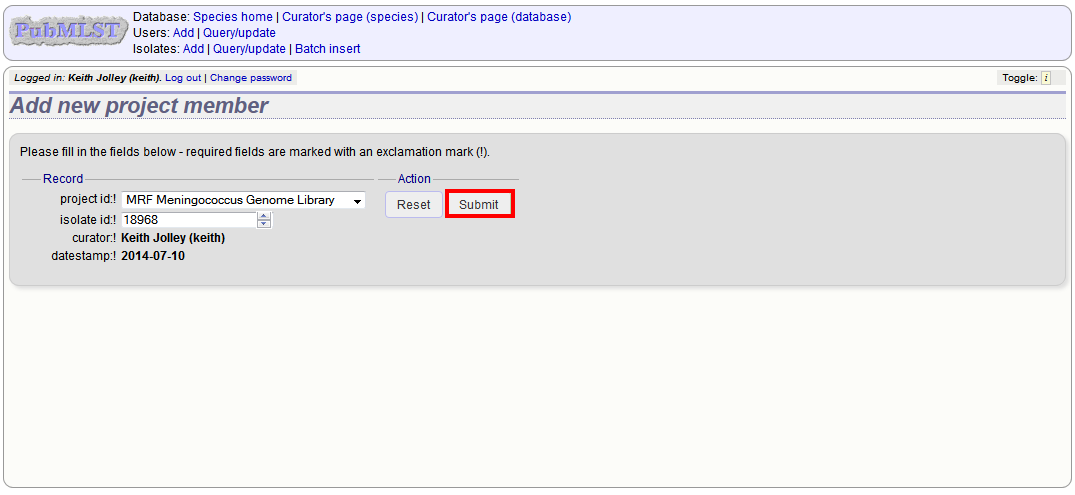
To add isolates in batch mode. Click the batch add (++) project members link on the curator’s main page.
Download an Excel submission template:
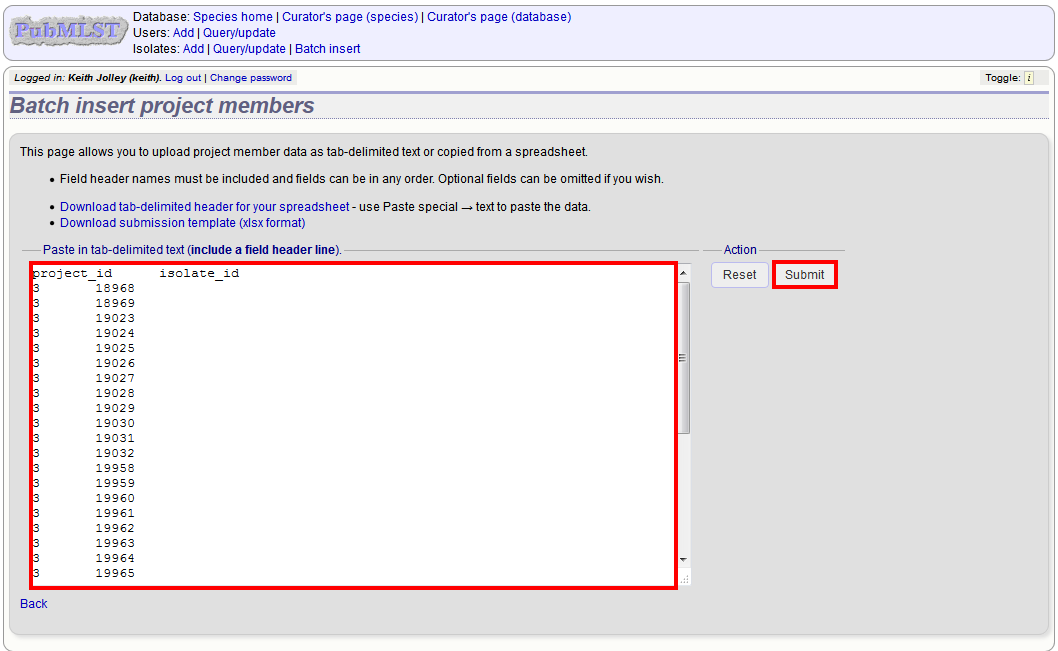
You will need to know the id number of the project - this is the id that was used when you created the project. Fill in the spreadsheet, listing the project and isolate ids. Copy and paste this to the web upload form. Press ‘Submit’.
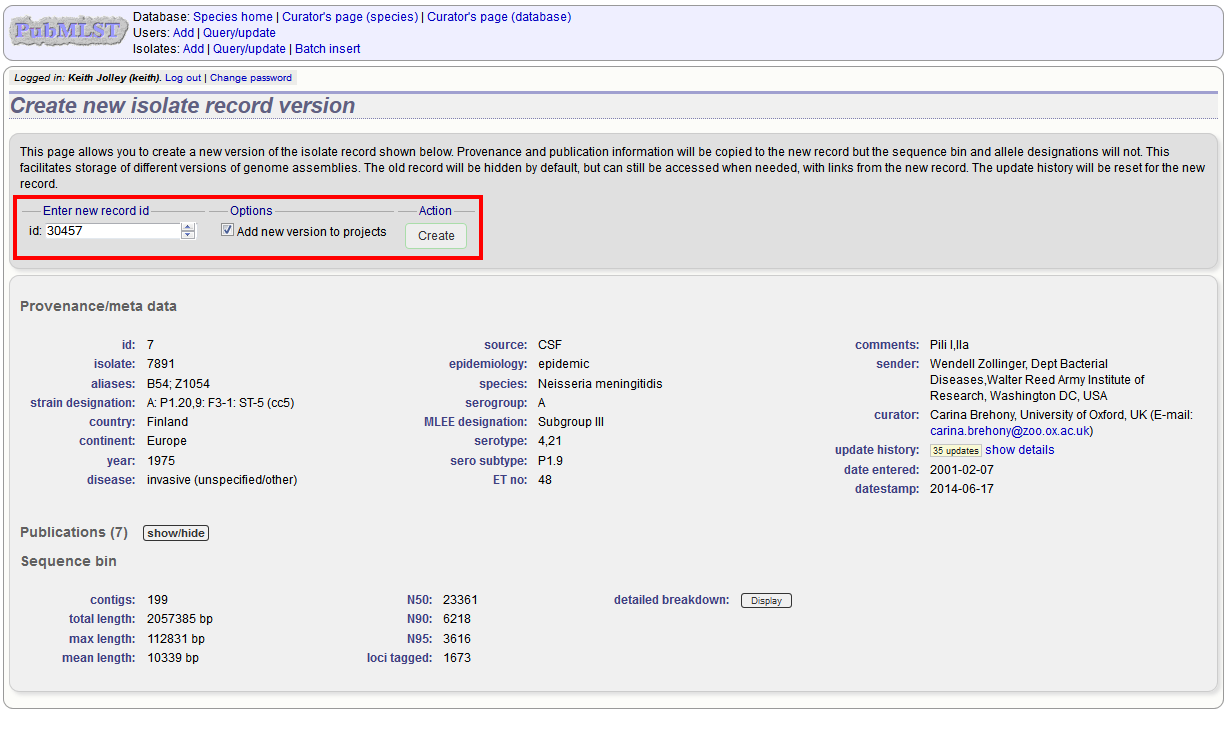
Isolate record versioning¶
Versioning enables multiple versions of genomes to be uploaded to the database and be analysed separately. When a new version is created, a copy of the provenance metadata, and publication links are created in a new isolate record. The sequence bin and allele designations are not copied.
By default, old versions of the record are not returned from queries. Most query pages have a checkbox to ‘Include old record versions’ to override this.
Links to different versions are displayed within an isolate record:
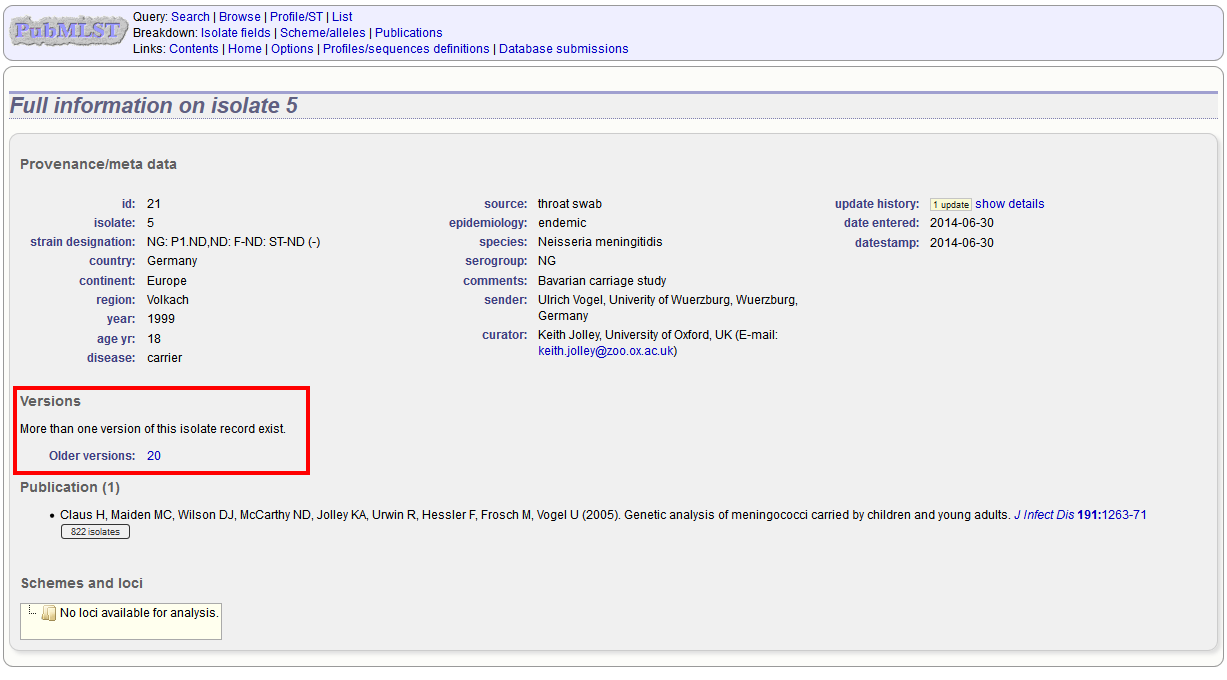
The different versions will also be listed in analysis plugins, with old versions identified with an [old version] designation after their name.
To create a new version of an isolate record, query or browse for the isolate:
Click the ‘create’ new version link next to the isolate record:
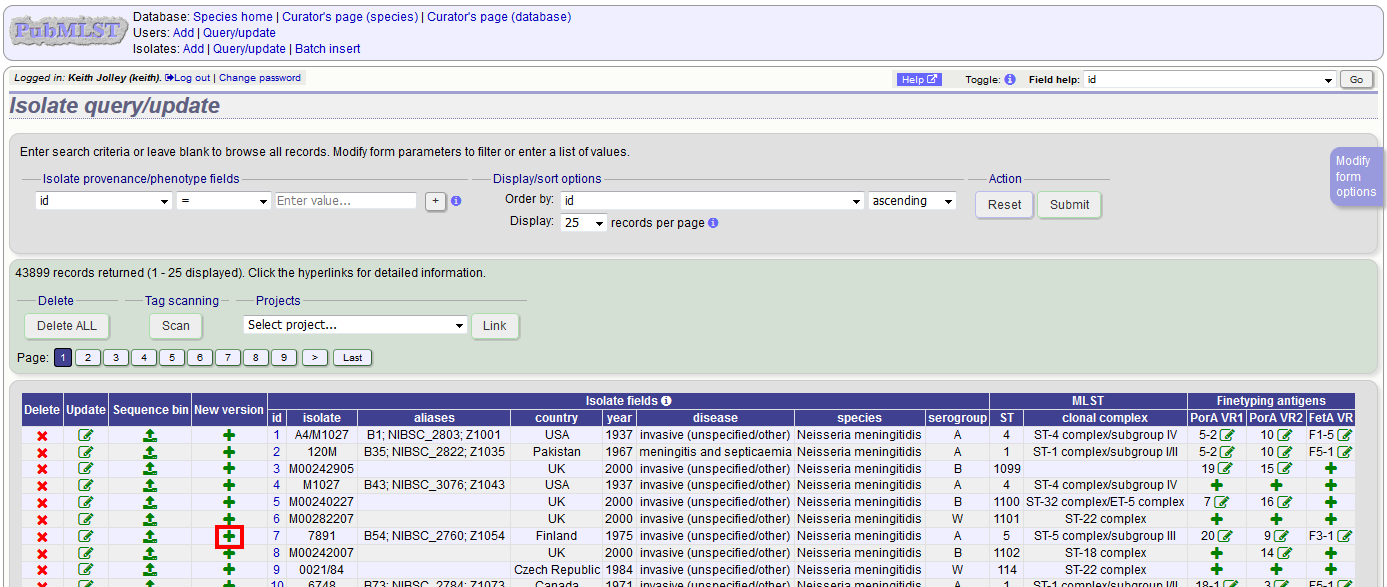
The isolate record will be displayed. The suggested id number for the new record will be displayed - you can change this. By default, the new record will also be added to any projects that the old record is a member of. Uncheck the ‘Add new version to projects’ checkbox to prevent this.
Click the ‘Create’ button.