BLAST
The BLAST plugin enables you to BLAST a sequence against any of the genomes in the database, displaying a table of matches and extracting matching sequences.
The function can be accessed by expanding the ‘Analysis’ section on the main contents page.
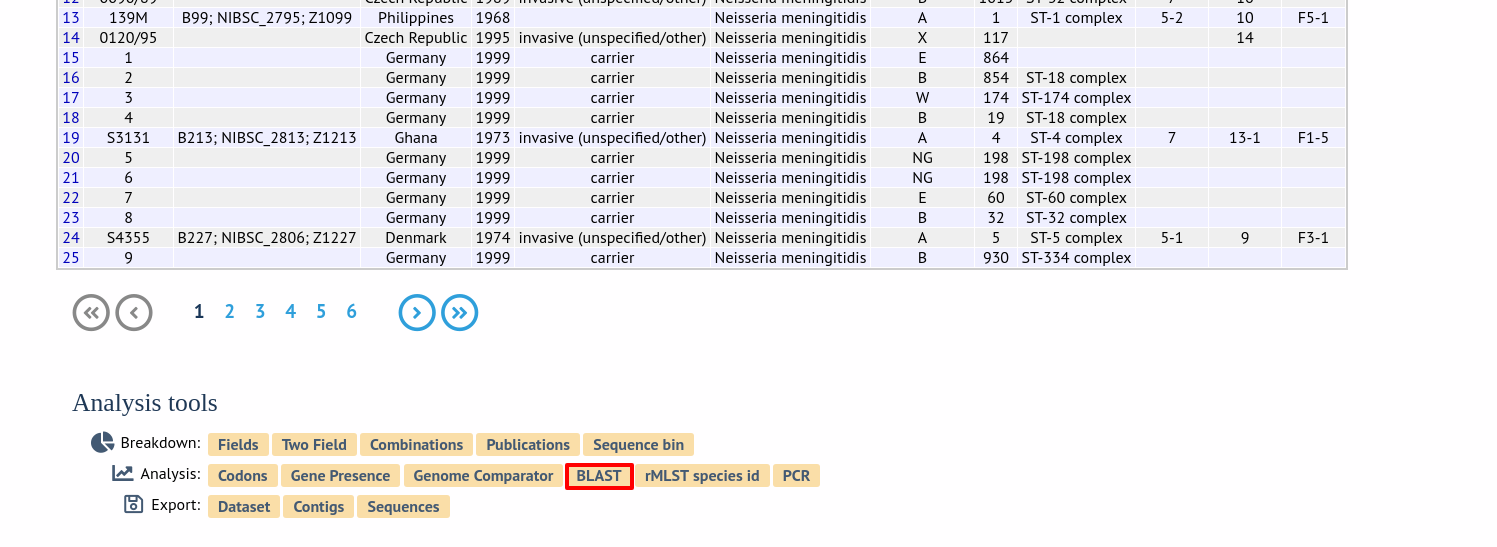
Alternatively,it can be accessed following a query by clicking the ‘BLAST’ button in the Analysis list at the bottom of the results table. Please note that the list of functions here may vary depending on the setup of the database.
Select the isolate records to analyse (on large databases you will need to enter a list of ids). These will be pre-selected if you accessed the plugin following a query. Paste in a sequence to query - this be either a DNA or peptide sequence.
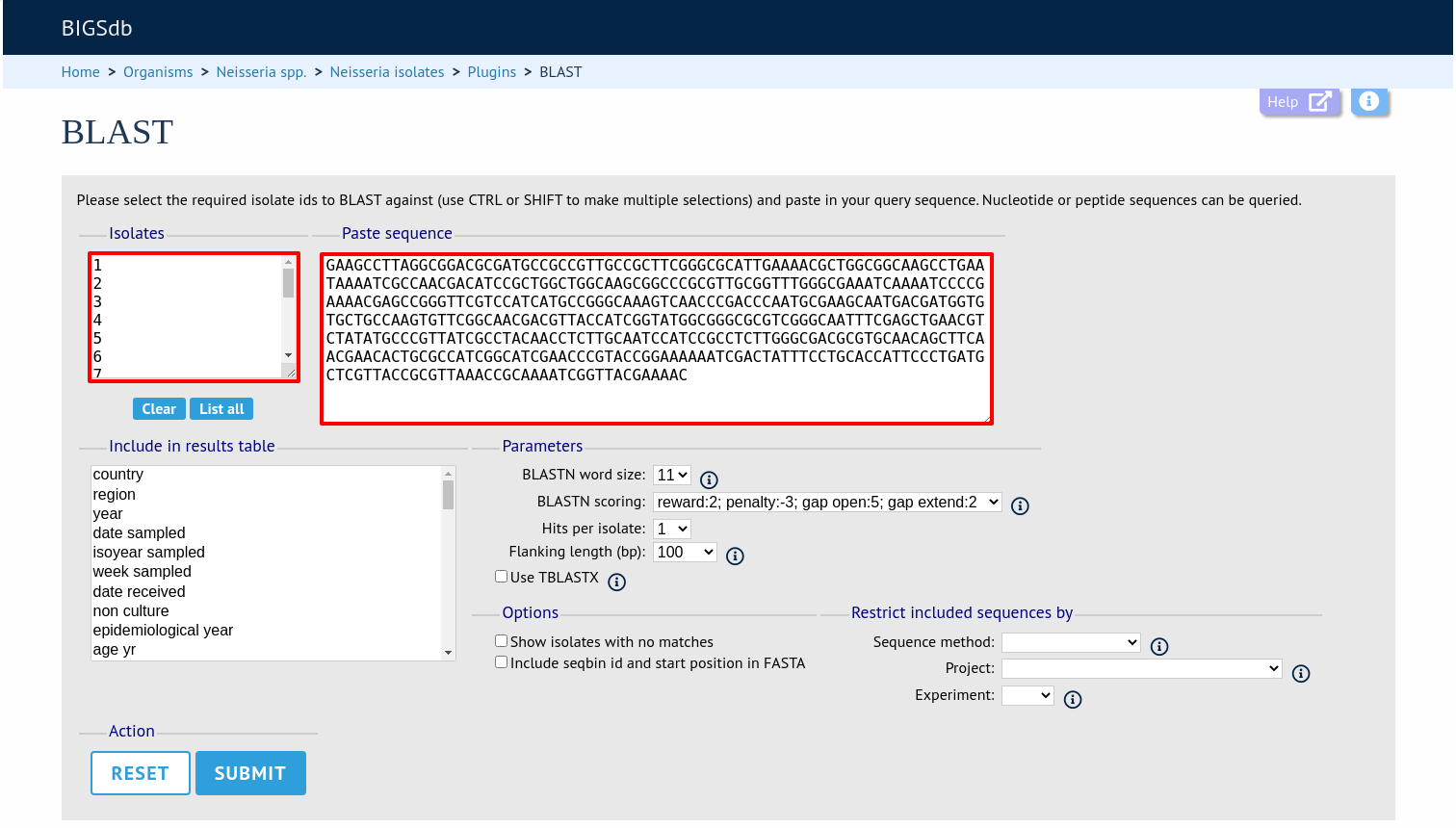
Click submit. If you are querying against 10 or fewer genomes then the results are run immediately, otherwise the job is sent to the job queue.
A table of BLAST results will be displayed.
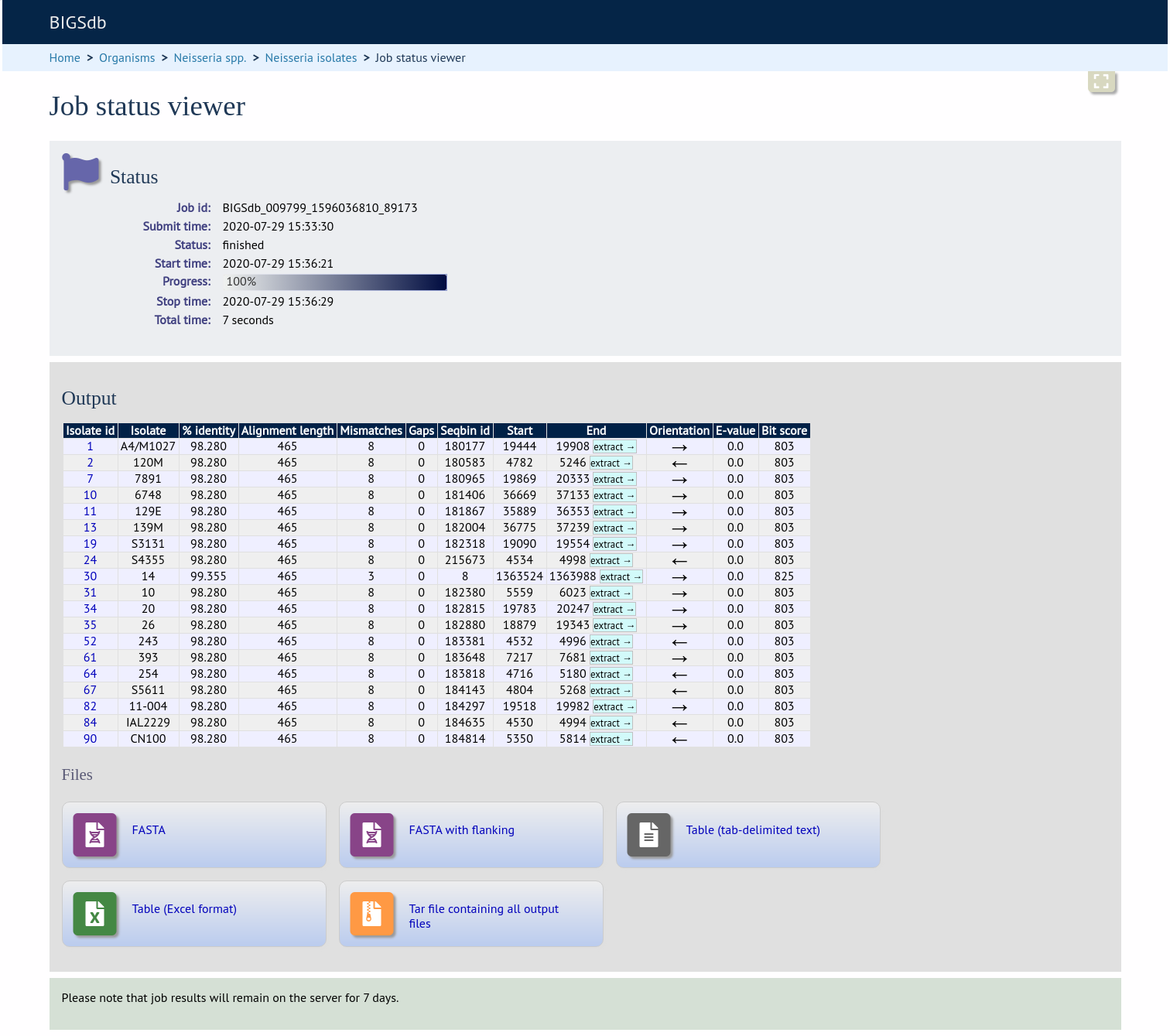
Clicking any of the ‘extract’ buttons to display the matched sequence.
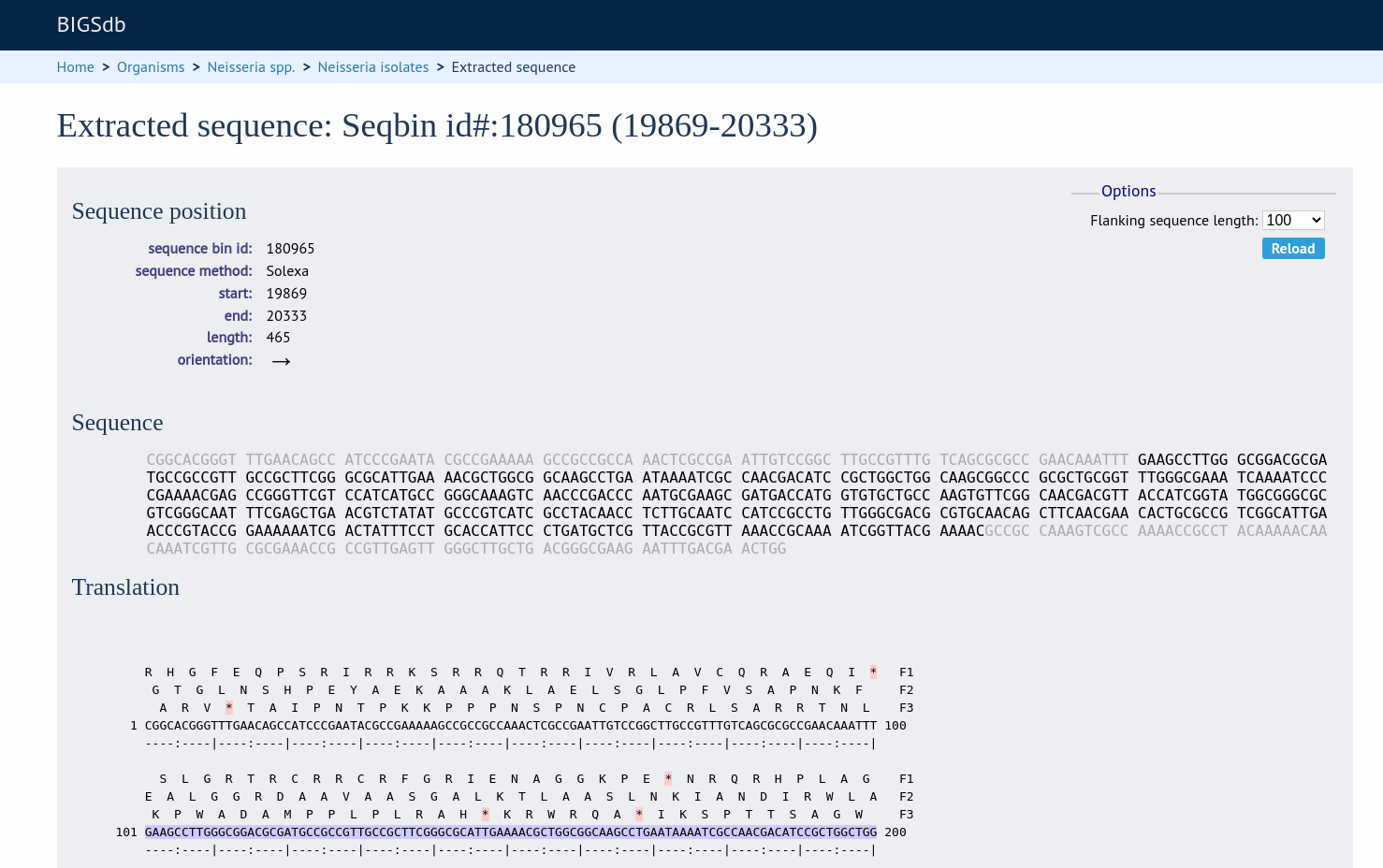
The extracted sequence is shown along with a translated sequence and flanking sequences.
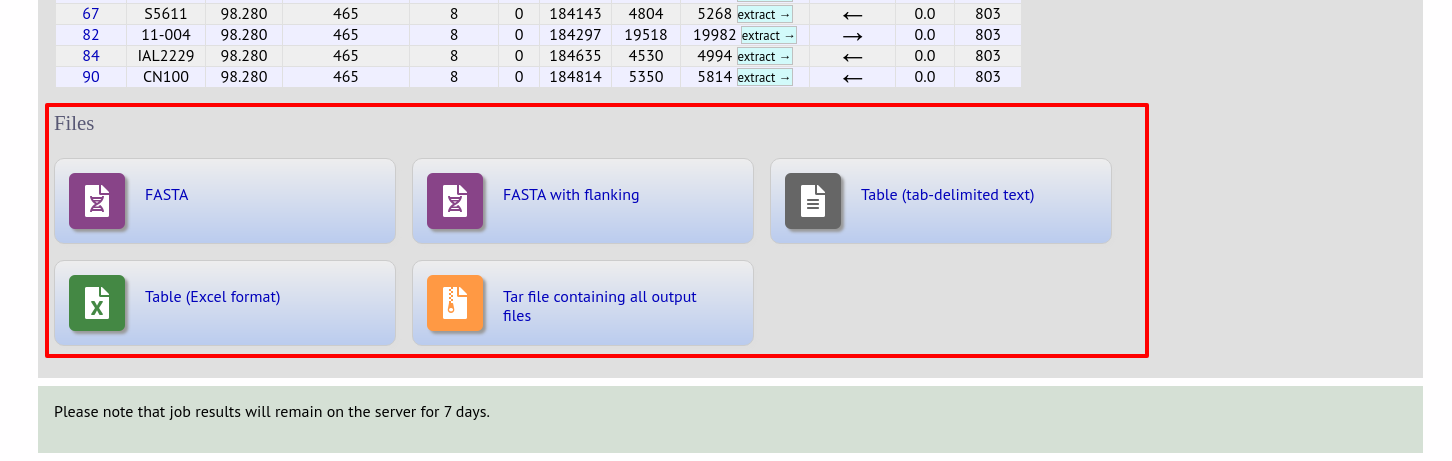
At the bottom of the results table are links to export the matching sequences in FASTA format, (optionall) including flanking sequnces. You can also export the table in tab-delimited text or Excel formats.
Include in results table fieldset
This selection box allows you to choose which isolate provenance fields will be included in the results table.
Multiple values can be selected.
Parameters fieldset
This section allows you to modify BLAST parameters. This affects sensitivity and speed.
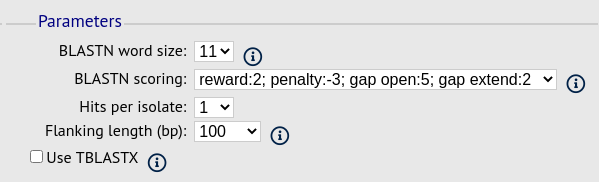
BLASTN word size - This is the length of the initial identical match that BLAST requires before extending a match (default: 11). Increasing this value improves speed at the expense of sensitivity.
BLASTN scoring - This is a dropdown box of combinations of identical base rewards; mismatch penalties; and gap open and extension penalties. BLASTN has a constrained list of allowed values which reflects the available options in the list.
Hits per isolate - By default, only the best match is shown. Increase this value to the number of hits you’d like to see per isolate.
Flanking length - Set the size of the upstream and downstream flanking sequences that you’d like to include.
Use TBLASTX - This compares the six-frame translation of your nucleotide query sequence against the six-frame translation of the contig sequences. This is significantly slower than using BLASTN.
No matches
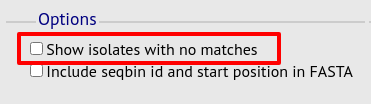
Click this option to create a row in the table indicating that a match was not found. This can be useful when screening a large number of isolates.
Filter fieldset
This section allows you to further filter your collection of isolates and the contig sequences to include.
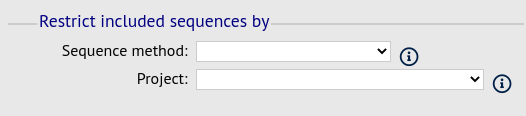
Available options are:
Sequence method - Choose to only analyse contigs that have been generated using a particular method. This depends on the method being set when the contigs were uploaded.
Project - Only include isolates belonging to the chosen project. This enables you to select all isolates and filter to a project.