Automated web-based sequence tagging
Sequence tagging, or tag-scanning, is the process of identifying alleles by scanning the sequence bin linked to an isolate record. Defined loci can either have a single reference sequence, that is defined in the locus table, or they can be linked to an external database that contains the sequences for known alleles. The tagging function uses BLAST to identify sequences and will tag the specific sequence region with locus information and an allele designation if a matching allele is identified by reference to an external database.
Select ‘scan’ sequence tags on the curator’s index page.

Next, select the isolates whose sequences you wish to scan against. Multiple isolates can be selected by holding down the Ctrl key. All isolates can be selected by clicking the ‘All’ button under the isolate selection list. On database with a large number of isolates, you will need to enter a list of isolate ids rather than pick from a list.
Select either individual loci or schemes (collections of loci) to scan against. Again, multiple selections can be made.
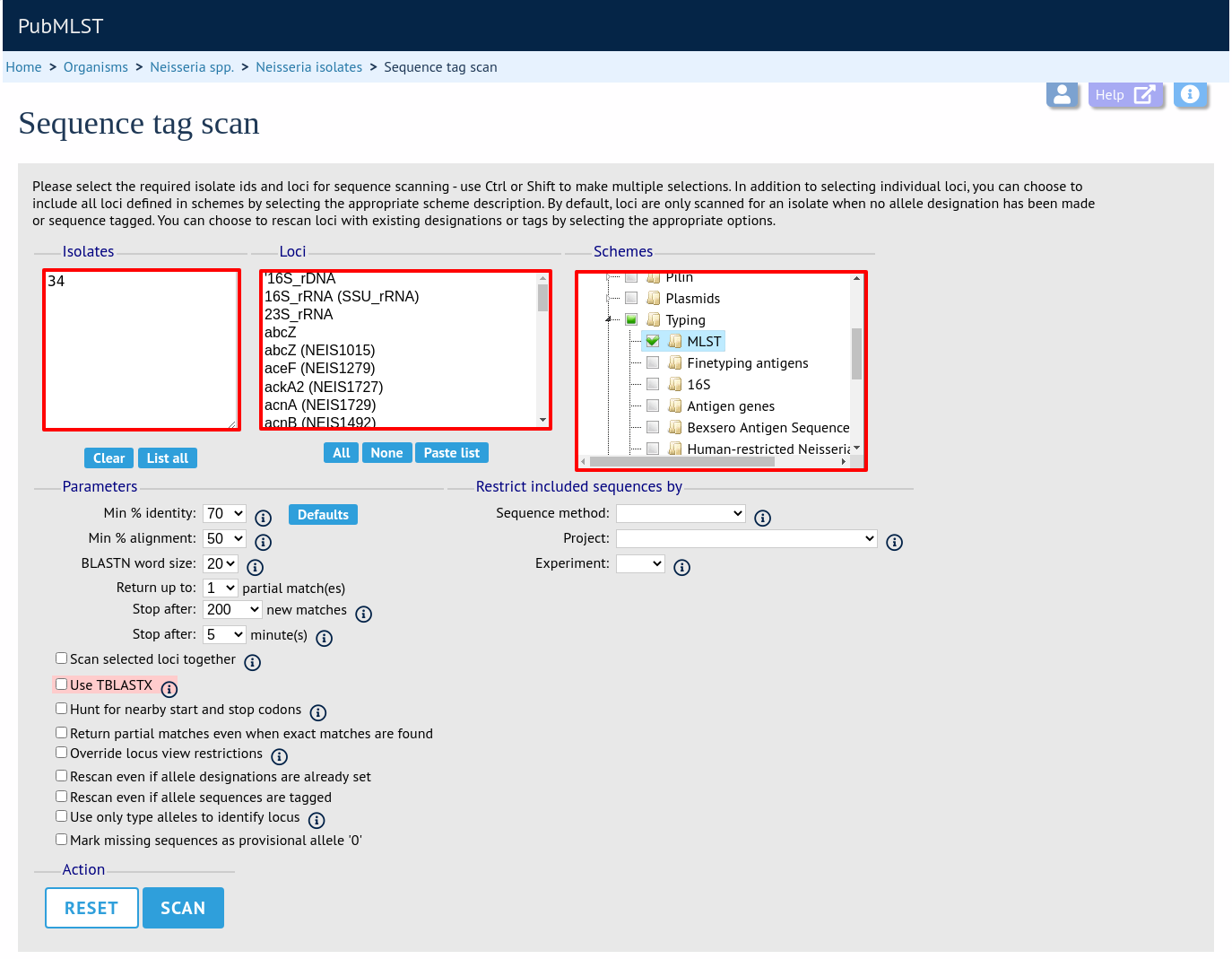
Choose your scan parameters. Lowering the value for BLASTN word size will increase the sensitivity of the search at the expense of time. Using TBLASTX (if it is available) is more sensitive but also much slower. TBLASTX can only be used to identify the sequence region rather than a specific allele (since it will only match the translated sequence and there may be multiple alleles that encode a particular peptide sequence).
By default, for each isolate only loci that have not had either an allele designation made or a sequence region scanned will be scanned again. To rescan in these cases, select either or both the following:
Rescan even if allele designations are already set
Rescan even if allele sequences are tagged
You can select to only use type alleles to identify the locus. This will constrain the search space so that allele definitions don’t become more variable over time. If a partial match is found to a type allele then a full database lookup will be performed to identify any known alleles. An allele can be given a status of type allele when defining.
If fast scanning is enabled, there will also be an option to ‘Scan selected loci together’. This can be significantly quicker than a locus-by-locus search against all alleles but is not enabled by default as it can use more memory on the server and requires exemplar alleles to be defined.
Options can be returned to their default setting by clicking the ‘Defaults’ button.

Press ‘Scan’. The system takes approximately 1-2 seconds to identify each sequence (depending on machine speed and size of definitions databases). Alternatively, if ‘Scan selected loci together’ is available and selected, it may take longer to return initial results but total time should be less (e.g. a 2000 loci cgMLST scheme may be returned in 1-2 minutes). Any identified sequences will be listed in a table, with checkboxes indicating whether allele sequences or sequence regions are to be tagged.
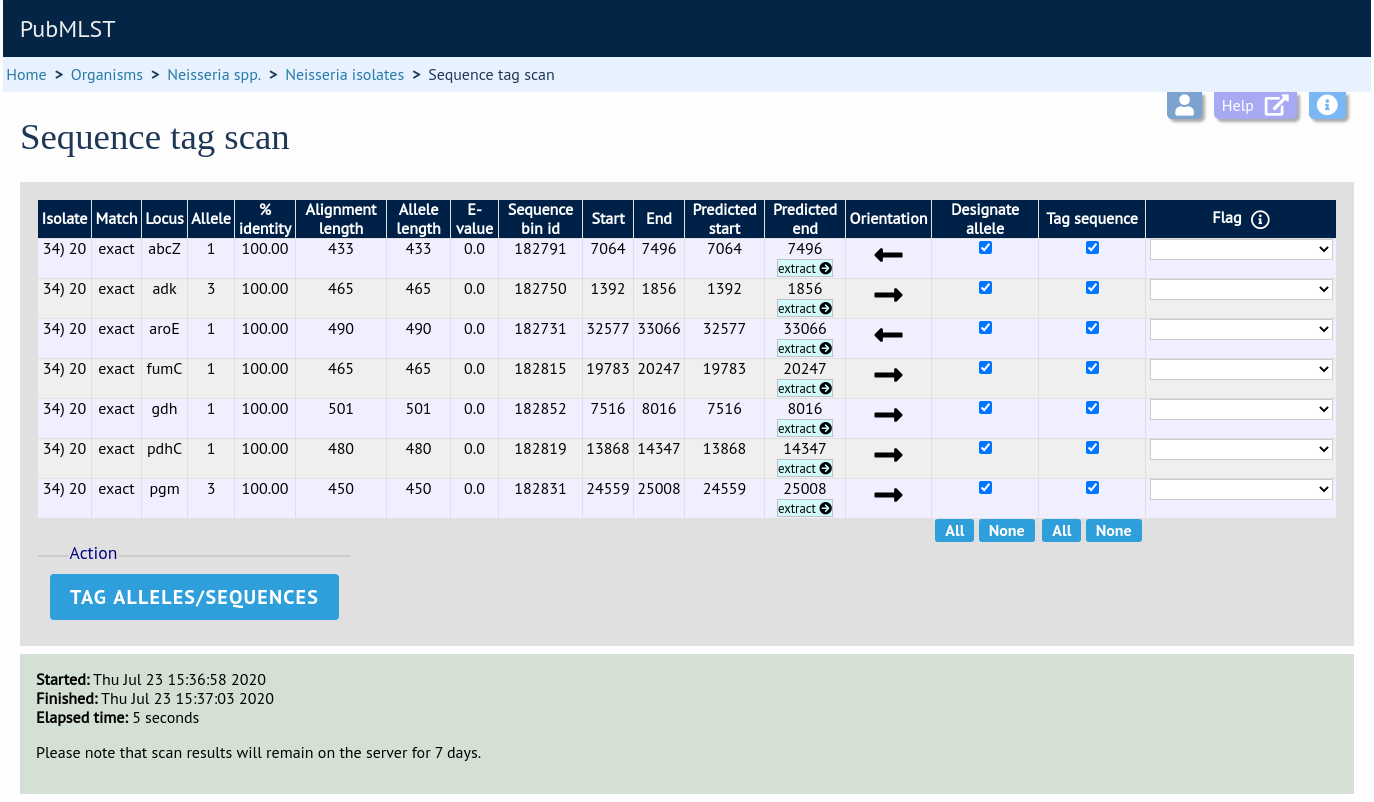
Individual sequences can be extracted for inspection by clicking the ‘extract →’ link. The sequence (along with flanking regions) will be opened in another browser window or tab.
Checkboxes are enabled against any new sequence region or allele designation. You can also set a flag for a particular sequence to mark an attribute. These will be set automatically if these have been defined within the sequence definition database for an identified allele.
See also
Ensure any sequences you want to tag are selected, then press ‘Tag alleles/sequences’.
If any new alleles are found, a link at the bottom will display these in a format suitable for automatic allele assignment by batch uploading to sequence definition database.